

Copyrights: Nur Ilyia Syazwani Saidin, Fatma Basyira Jamallodin, Mohd Nazri Hassan, Salfarina Iberahim, Abdul Hanan Abdullah, Muhamad Aidil Zahidin, Zefarina Zulkafli, Noor Haslina Mohd Noor, 2024. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Introduction: A deficiency in antithrombin (AT) can be hereditary or acquired. It is characterized by an AT activity level that is less than 80% of normal or the lower limit of the reference range on a regular basis. In some cases, AT deficiency has been linked to an increased risk of thromboembolism.
Case presentation: We present the case of a 56-year-old Malay man with long-segment thrombosis of the portal vein and superior mesenteric vein with small bowel ischemia. He was diagnosed with AT deficiency following an extensive hematological and thrombophilia workup supported by a strong familial history of venous thromboembolism (VTE) affecting his brothers and sister.
Conclusion: Inherited AT deficiency must be considered when spontaneous VTE occurs in young patients with unusual localizations, such as mesenteric veins and portal veins.
INTRODUCTION
Antithrombin (AT) is a serpin and natural anticoagulant that functions predominantly by inactivating the coagulation factors thrombin and factor Xa and less effectively by inactivating the factors IXa and XIa1, 2 Furthermore, the anticoagulant heparin is dependent on AT for its activity. Heparin binds to AT and increases its anticoagulant activity one thousandfold. AT, like tissue factor (TF) pathway inhibitor, inhibits the TF–factor VIIa complex, preventing factor VIIa from activating factors IX and X1.
AT deficiency is an autosomal dominant type of thrombophilia affected by serpin family C member 1 (SERPINC1) gene mutations3 and is reported to affect 1 in 500 to 1 in 5,000 people2, 4. Among Asians, 0.15% of individuals in healthy Japanese populations were estimated to have AT deficiency, which is comparable to the rate in Caucasian populations5. Individuals with AT deficiency are more prone to thrombosis. It is expected that 50% of those with congenital AT deficiency will have developed venous thromboembolism (VTE) by the age of 506. Homozygous AT deficiency is uncommon compared to heterozygous AT deficiency, and most homozygosity has been linked to intrauterine death2.
This case report describes a 56-year-old man with heterozygous AT deficiency who presented with acute abdominal pain due to long-segment thrombosis of the portal vein and superior mesenteric vein and small bowel ischemia.
CASE PRESENTATION
A 56-year-old Malay man presented to the emergency department with a sudden onset of lower abdominal pain that continued for several hours. The pain initially started in the left testicular area and radiated to the left lower quadrant of the abdomen. The pain was extensive and described by the patient as 10/10 on the pain grading scale. The abdominal pain was associated with a fever. He had no other symptoms such as nausea, vomiting, diarrhea, dysuria, or hematuria. He denied any traditional medication or recreational drug use. He used to be a cigarette smoker but had quit for the past 10 years.
The patient had a history of leg swelling 20 years ago but was unsure about the diagnosis. He had no further follow-up and received no further treatment. He had a strong family history of hypercoagulability, as his elder brother had a stroke, and his younger brother and sister had deep vein thrombosis (DVT) and were on anticoagulant treatment. The results of the thrombophilia workups for these patients were not found; they were probably treated at another hospital.
The physical examination revealed that he was afebrile with normal vital signs. The rest of the examination was unremarkable except for generalized lower abdominal tenderness but no guarding or rebound.
The laboratory blood tests reveal the following: a total white cell count of 15.17 × 109/L, a hemoglobin level of 12.7 g/dL, and a platelet count of 413 × 109/L. The coagulation test showed normal prothrombin time and activated partial thromboplastin time (12.9 seconds and 36.0 seconds, respectively). He had normal liver and renal function. The autoimmune screening tests, including for antinuclear antibody, complement levels (C3 and C4), rheumatoid factor, and anticardiolipin antibody, were negative.
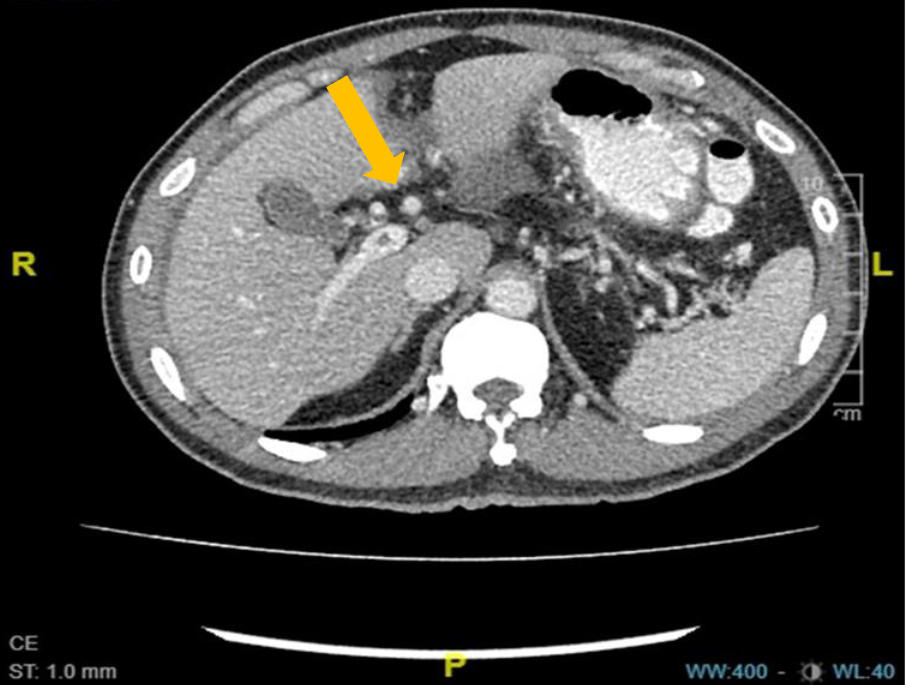
An ultrasound scan of the patient’s abdomen showed free fluid in the subhepatic space, right iliac fossa, and Morrison’s pouch. A computed tomography scan of the abdomen performed on a subsequent day showed long-segment thrombosis of the portal vein and superior mesenteric vein with small bowel ischemia (Figure 1). He was then treated with subcutaneous fondaparinux for 10 days and showed improvement. He was discharged to his home with a daily oral warfarin dose of 3 mg. He was referred for hemophilia workup during subsequent outpatient follow-up.

A thrombophilia screening was conducted during the subsequent outpatient clinic follow-up (12 months after the thrombotic events) (Table 1). The screening revealed that the patient had persistently low levels of AT. Based on his clinical presentation and laboratory findings, he was diagnosed with a heterozygous deficiency of AT. He was then scheduled for life-long anticoagulant therapy for thromboprophylaxis.
He was on 3 mg of warfarin daily with regular international normalized ratio (INR) monitoring. He aimed for an INR between 2 and 3. However, he was having difficulty maintaining the INR within the targeted therapeutic range. He was prescribed rivaroxaban, an orally active direct factor Xa inhibitor. However, rivaroxaban was discontinued after one month due to an inability to tolerate the side effect of epigastric pain. The warfarin was restarted, and currently he is taking a daily 2 mg tablet of warfarin. He was compliant with the treatment, and no other thrombotic or bleeding events have been documented.
| Test | Result | Reference range | |
|---|---|---|---|
| July 2017 | September 2017 | ||
| Protein C activity | 125% | 128% | 70–140% |
| Free protein S antigen | 92% | 92% | 72–123% |
| AT activity | 46% | 52% | 83–128% |
| APCR V Leiden | 2.94 | 3.2 | Normal ratio > 2.1 |
DISCUSSION AND CONCLUSION
AT deficiency was discovered in 1965 by Olav Egeberg in a Scandinavian family with VTE. He also determined that the deficiency is an autosomal dominant disorder7.
AT is a heparin cofactor and a member of the serine protease inhibitor family. The mature AT molecule consists of 432 amino acids and is mainly produced in the liver. AT is a thrombin and factor Xa protease inhibitor. However, AT also has the ability to inhibit factors IXa, XIa, and XIIab; kallikrein; and plasmin8.
Normal AT plasma levels range from 112–140 μg/mL, with normal AT antigen and activity levels in the range of 80–120%. AT levels are lower in newborns than in adults and somewhat lower in older men and in women who take birth control tablets4.
Given that AT is one of the major naturally occurring coagulation inhibitors, acquired or inherited deficiencies in this protein result in increased thrombin production8. Most of the inherited AT deficiencies are heterozygous. Homozygosity is uncommon and has an increased risk of mortality during pregnancy7. In the general population, the incidence has been estimated as 1 in 20 to 1 in 200 people9. Meanwhile, 1 in 600 people are born with a congenital AT deficiency4.
In type I or quantitative deficiency, no abnormal AT is observed in plasma, and the plasma AT ratio of antigen to anticoagulant activity is near 110. Short deletions and insertions are the most common causes of type I AT deficiencies7.
In type II or qualitative deficiency, a high concentration of AT variants with impaired or null anticoagulant activity is detected in plasma. The risk of thrombosis in patients with type II deficiency is very heterogeneous10. Type II AT deficiencies are commonly caused by single base pair substitutions in the reactive domain (type IIa) or in the heparin-binding region of AT (type IIb), resulting in qualitative abnormalities7. Mayu et al.11 demonstrated that FS-3Stop and Met32Thr mutations cause type I AT deficiency, whereas Ser116Pro and Ala59Val mutations cause type II AT deficiency, demonstrating the existence of multiple molecular pathways underlying AT deficiency.
Unprovoked DVT, with or without complicated pulmonary embolism, is the most common manifestation observed (approximately 75% of incidents). However, thrombosis may also occur more frequently in atypical venous territories, such as the splanchnic veins (including Budd–Chiari syndrome), vena cava system, and cerebral sinuses10.
The goal of treatment for patients with inherited AT deficiency is to enhance AT activity (to 120% of normal levels) and maintain AT activity (to 80% of normal levels). Treatment options for those with inherited AT deficiency include plasma-derived AT, heparin, fresh frozen plasma, and human recombinant AT8. The first line of care for VTE in patients with AT deficiency is often the same as that for any other patient: (i) consideration of thrombolytics, (ii) initial therapy with heparin or fondaparinux, and (iii) a transition to a vitamin K antagonist. When evaluating individuals with extensive or clinically symptomatic VTE, it may be relevant to consider AT concentration7.
The most commonly used oral anticoagulants in the treatment of AT deficiency are vitamin K antagonists because they have the most clinical experience and are universally approved in this setting. Direct oral anticoagulants (DOACs), on the other hand, have only recently been offered as a novel and promising therapeutic. DOACs are increasing in popularity, and their use should be considered in patients who have AT deficiency, as these medications work via an AT-independent mechanism10.
Abbreviations
AT: Antithrombin, VTE: Venous Thromboembolism, DVT: Deep Vein Thrombosis, SERPINC: Serpin Family C Member 1 (Gene), TF: Tissue Factor, INR: International Normalized Ratio, APCR: Activated Protein C Resistance
Acknowledgments
None.
Author’s contributions
Research concept and design: Noor Haslina Mohd Noor, Nur Ilyia Syazwani Saidin, Fatma Basyira Jamallodin; Collection and processing of material: Mohd Nazri Hassan, Abdul Hanan Abdullah; Text writing: Noor Haslina Mohd Noor, Muhamad Aidil Zahidin; Editing: Zefarina Zulkafli; Approval of the manuscript final version: Salfarina Iberahim, Noor Haslina Mohd Noor. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
This study was carried out in line with the revised Helsinki Declaration. The study was authorised by the institutional review board, and a participant provided informed consent.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
References
-
Kyotani
M.,
Okumura
K.,
Takagi
A.,
Murate
T.,
Yamamoto
K.,
Matsushita
T.,
Molecular basis of antithrombin deficiency in four Japanese patients with antithrombin gene abnormalities including two novel mutations. American Journal of Hematology.
2007;
82
(8)
:
702-5
.
View Article PubMed Google Scholar -
Bravo-Pérez
C.,
de la Morena-Barrio
M.E.,
Vicente
V.,
Corral
J.,
Antithrombin deficiency as a still underdiagnosed thrombophilia: a primer for internists. Polish Archives of Internal Medicine.
2020;
130
(10)
:
868-77
.
View Article PubMed Google Scholar -
Wells
P.S.,
Blajchman
M.A.,
Henderson
P.,
Wells
M.J.,
Demers
C.,
Bourque
R.,
Prevalence of antithrombin deficiency in healthy blood donors: a cross-sectional study. American Journal of Hematology.
1994;
45
(4)
:
321-4
.
View Article PubMed Google Scholar -
Rodgers
G.M.,
Role of antithrombin concentrate in treatment of hereditary antithrombin deficiency. An update. Thrombosis and Haemostasis.
2009;
101
(5)
:
806-12
.
View Article PubMed Google Scholar -
Patnaik
M.M.,
Moll
S.,
Inherited antithrombin deficiency: a review. Haemophilia.
2008;
14
(6)
:
1229-39
.
View Article PubMed Google Scholar -
Fischer
R.,
Sachs
U.J.,
Heidinger
K.S.,
Eisenburger
D.,
Kemkes-Matthes
B.,
Prevalence of hereditary antithrombin mutations is higher than estimated in patients with thrombotic events. Blood Coagulation & Fibrinolysis.
2013;
24
(4)
:
444-8
.
View Article PubMed Google Scholar -
Sakata
T.,
Okamoto
A.,
Mannami
T.,
Matsuo
H.,
Miyata
T.,
Protein C and antithrombin deficiency are important risk factors for deep vein thrombosis in Japanese. Journal of Thrombosis and Haemostasis.
2004;
2
(3)
:
528-30
.
View Article PubMed Google Scholar -
Tait
R.C.,
Walker
I.D.,
Perry
D.J.,
Islam
S.I.,
Daly
M.E.,
McCall
F.,
Prevalence of antithrombin deficiency in the healthy population. British Journal of Haematology.
1994;
87
(1)
:
106-12
.
View Article PubMed Google Scholar -
G\uaman
A.M.,
G\uaman
G.D.,
Deficiency Of Antithrombin III (AT III) - Case Report and Review of the Literature. Current Health Sciences Journal.
2014;
40
(2)
:
141-3
.
PubMed Google Scholar -
Wan Ab Rahman
W.S.,
Abdullah
W.Z.,
Hassan
M.N.,
Hussin
A.,
Zulkafli
Z.,
Haron
J.,
Familial antithrombin III deficiency in a Malay patient with massive thrombosis. The Malaysian Journal of Pathology.
2017;
39
(2)
:
197-200
.
PubMed Google Scholar -
Roberts
J.C.,
von Drygalski
A.,
Zhou
J.Y.,
Rodgers
G.M.,
Ansteatt
K.,
Tarantino
M.D.,
Five Challenging Cases of Hereditary Antithrombin Deficiency Characterized by Thrombosis or Complicated Pregnancy. Journal of Blood Medicine.
2022;
13
:
611-8
.
View Article PubMed Google Scholar
Comments
Article Details
Volume & Issue : Vol 11 No 2 (2024)
Page No.: 6179-6182
Published on: 2024-02-29
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
- HTML viewed - 3140 times
- PDF downloaded - 1099 times
- XML downloaded - 109 times
