

Copyrights: Nikhil Choudhary, Kalyani Raju, Arun Heddur Shanthappa, CSB Rajendra Prasad, Sandesh Agarwal, 2022. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Myeloma is the neoplastic proliferation of plasma cells. The peak age of onset of this disease is the seventh decade, and it is rare in young patients. Myeloma usually presents as lytic lesions in axial bones and rarely present as a joint disease. Usually, the tumor cells secrete monoclonal immunoglobulins. Non-secretory myeloma is a rare clinical entity. The incidence of light-chain myeloma is 16 - 20 % among all multiple myelomas, and the more common subtype is the lambda chain type. We present a case of kappa light chain-type myeloma in a 29-year-old male with the presenting lesion in the right knee joint. The case is presented for its rarity due to the age of patient, presentation, and type of myeloma.
Introduction
Multiple myeloma (MM) is defined as a neoplastic proliferation of plasma cells derived from a single cell lineage (B-cell) in the marrow that characteristically secretes an abnormal immunoglobulin, leading to monoclonal gammopathy. The monoclonal immunoglobulins and most commonly IgG or IgA. The protein is mostly represented by an intact immunoglobulin (heavy and light chain), or it can be represented only by a light chain1.
The peak age of onset for MM is the seventh decade, and it is rare in young patients. It is more commonly reported in males than females2, 3. MM commonly affects axial bones and can rarely manifest as joint disease2. It causes end-organ damage, which can manifest as hematological, bone, or renal complications.
MM can present in a premalignant phase where clonal plasma cells are present without end-organ damage. Non-secretory myeloma is a rare clinical entity of MM with proliferation of monoclonal plasma cells of the bone marrow; however, the plasma cells do not secrete immunoglobulin. Light-chain myeloma (LCM) is rare; plasma cells secrete only light chains, and it can be either lambda or kappa light chain type3.
We present a case of light-chain MM (LCMM) in a young male.
Case Report
A 29-year-old male patient presented with pain and swelling in the right knee joint for 3 months, with insidious onset. The pain was dull aching, progressive in nature, worse in the morning, and interfered with his daily activities. There was no history of fever, loss of weight, loss of appetite, generalized weakness, headache, or hemorrhagic tendencies. Past history and personal history were not significant. There was no history of drug intake for long duration.
General physical examination was within normal limits. Local examination of the right knee joint revealed swelling, increased temperature, and tenderness. Swelling measured approximately 5×3 cm over the distal thigh (medial femoral condyle). Restriction of movement was noted. Other bony prominences were palpable. Ankle and toe movement was normal. Pulse and sensation distal to the knee joint were normal. Systemic examination was within normal limits.



Hematological investigation revealed hemoglobin 12 gm/dl, red blood cell (RBC) count 3.65 million/cu mm, PCV 34%, platelet count 1.35 lakh/mm3, and ESR 35 mm/hour. White blood cell (WBC) parameters were within normal limits. Peripheral blood smear showed a normocytic normochromic blood picture. Liver function tests (LFTs) were within normal limits, except for serum globulin, which was 2.70 g/dl. Renal function test (RFT), serum electrolytes, and serum calcium were within normal limits. C-reactive protein (CRP) was negative.
Radiograph of the right knee joint showed lytic lesions in the lower part of femur and upper end of tibia and fibula (Figure 1 a). Skull X-ray showed lytic lesions (Figure 1 b). X-ray of the left humerus showed lytic lesions (Figure 1 c). Magnetic resonance imaging (MRI) of the right knee joint showed a hyperintense lesion with its epicenter in the medullary space of the meta-epiphyseal region of distal femur and proximal tibia and fibula.
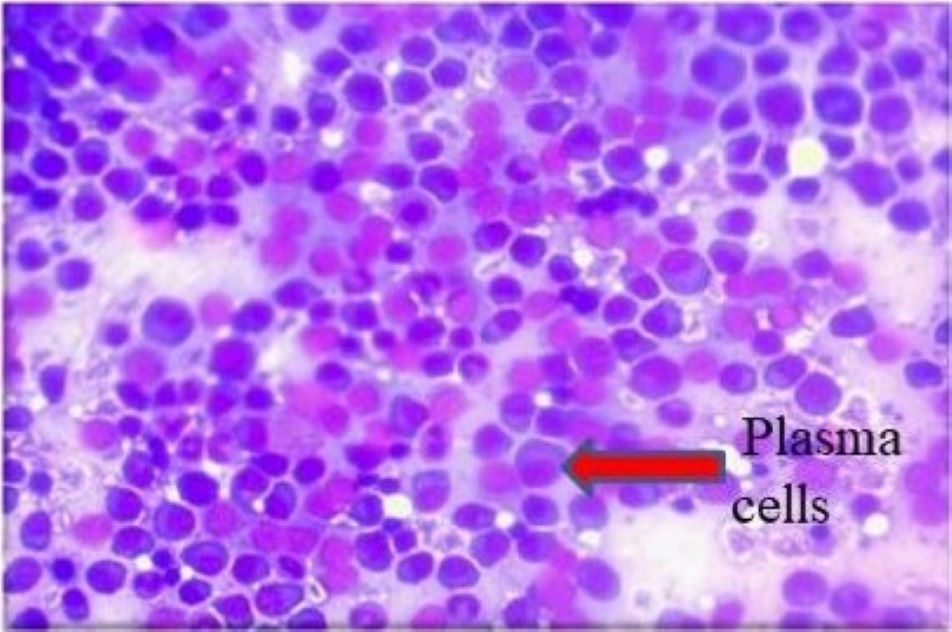
Fine-needle aspiration (FNA) of the right distal femur yielded scanty hemorrhagic material. Microscopy showed a highly cellular smear; cells were discohesive, arranged in singles, and had features of plasma cells (Figure 2). The cytological features were suggestive of a plasma cell disorder. Tru-Cut biopsy of the swelling in the right distal femur was performed. Grossly, the tissue was multiple, linear, grey-white to grey-brown, soft tissue bits that altogether measured 1×0.5 cm. Microscopy revealed fragments of fibro-collagenous tissue and plasma cells in sheets, similar to the FNA smears. The features of the Tru-Cut biopsy were suggestive of a plasma cell disorder. Urine for Bence–Jones protein was positive.
The blood sample was subjected to serum protein electrophoresis and was normal; no M-spike was noted on electrophoresis (Figure 3). Bone marrow aspiration of the right posterior superior iliac spine was performed. It showed hypercellular marrow, and the myeloid-erythroid ratio was 2:1. Cells of erythroid series showed normal maturation. The differentials of cells of myeloid series were: promyelocytes 4%, myelocytes 5%, metamyelocytes 0%, neutrophils 10%, lymphocytes 5%, eosinophils 0%, band forms 15%, and plasma cells 61%. Megakaryocytes were normal in number and morphology. Therefore, the bone marrow study showed features consistent with plasma cell disorder.
Free light-chain assay (FLCA) was performed with a blood sample and showed free kappa light chains of 16,100 mg/L (normal range 3.3 – 14.40 mg/L) and free lambda chains 7.80 mg/L (normal range 5.76 – 26.30 mg/L), with a kappa-lambda ratio of 2064.103 (Normal range 0.26 – 1.65).
Based on the microscopic features of the FNA smears, Tru-Cut biopsy findings, bone marrow aspiration smears, and the presence of Bence–Jones protein in the urine, as per the Durie and Salmon criteria4, a diagnosis of MM was proposed. Considering the markedly increased levels of serum free kappa light chains and elevated kappa-to-lambda ratio, a final diagnosis of LCM/Bence–Jones myeloma was proposed.
Discussion
Myeloma is a hematological malignancy involving monoclonal plasma cell proliferation that invades the bone marrow. The annual incidence of MM is 7.74 per 100,000 population4, 5, 6. It is twice as common in African Americans than in Caucasians. MM occurs most often in people over the age of 60 years. The age at diagnosis is usually 70 years. About 2% of cases occur in people under 40 years of age. MM has a slight male predilection. Risk factors for MM include exposure to radiation, chemicals, asbestos, benzene, pesticides, and chemicals used in the rubber industry and wooden products in carpentry. People with a history of a solitary plasmacytoma of the bone are at higher risk for developing MM7. Patient with a low concentration of M protein in the serum have a 1 – 2% chance of developing myeloma8. In the present case, the patient was a 29-year-old male with no history of exposure to risk factors or any predisposing disorders.
MM is usually preceded by a premalignant stage known as monoclonal gammopathy of undetermined significance (MGUS). MGUS presents with detection of M-protein without a plasma cell or lymphoproliferative disorder. The features of MGUS are: M-protein < 3 g/dL, less than 10% plasma cells in bone marrow, absence or small quantity of M-protein in the urine, absence of lytic bony lesions, anemia, hypercalcemia, renal insufficiency, and stability of M-protein over time. The majority of cases of MM initiate from non-IgM MGUS, and 20% of cases initiate from light-chain MGUS4. In the present case, plasma cells were >60% in bone marrow aspiration, and lytic lesions were noted on skull X-ray.
MM typically presents with bone disease including bone pain, pathological fractures, spinal cord compression, renal function impairment, anemia, hypocalcemia, persistent bacterial infection (usually Streptococcus pneumonia), and generalized weakness. The most common among these presentations is bone pain, and it is seen in approximately 70% of patients presenting with lower back pain. MM can rarely manifest as joint disease; 30% of cases manifest with pathological fracture and 20% with renal failure. Approximately 30% of MM cases are detected incidentally during routine blood investigations2, 7. The patient in the present case presented with pain and swelling of the right knee joint in addition to mild thrombocytopenia, mildly elevated ESR, and without characteristics rouleux formation on peripheral blood smear. Serum calcium and creatinine levels were normal.
MM distinctively involves the presence of clonal plasma cells in the marrow that characteristically secrete an abnormal immunoglobulin (most commonly IgG followed by IgA), leading to monoclonal gammopathy. The serum protein is represented by an intact immunoglobulin (heavy and light chain), or it can be represented by only a light chain1. Serum protein electrophoresis shows a narrow peak most commonly in the region of γ-globulin in case of secreting myelomas4. The presence of only a light-chain monoclonal protein is observed in 20% of myeloma cases and is called as LCM. Around 1 — 2% of patients have non-secretory MM with no evidence of M protein6. In the present case, serum electrophoresis did not indicate the presence of M protein; however, FLCA indicated the presence of light chain.
Non-secretory myeloma (NSM) is a rare variant and constitutes about 1% of myelomas. It usually presents with bone pain and bone lesions. Anemia may be a presenting feature. Serum and urine monoclonal proteins are absent. Renal failure and increased calcium levels are generally not seen. Bone marrow biopsy must be performed in suspected cases followed by immunostaining on biopsy sections, which may establish the diagnosis6. Two characteristic types of NSM have been described. In the first type, patients are non-producers. In these patients, tumors have a defect in immunoglobulin synthesis and are unable to synthesize protein, even though they have features of a plasma cell disorder. The other group of non-secretory myeloma patients have tumors that produce a protein with defective secretion3.
The incidence of LCM is about 16 — 20% worldwide among all MM patients6. LCMM has an early age of onset. In patients diagnosed with LCMM, plasma cells show rearrangements in the immunoglobulin heavy chain (IgH) at the DNA level; as a result, they are unable to produce IgH. One IgH allele has a germ-line configuration (for the D, J, and C domains), and the second allele participates in translocation. These features contrast those of classical MM, where one allele has a functional rearrangement, and the other allele is usually involved in translocation. LCM is a malignant disease involving unusual plasma cell proliferation and secretion of free light chains (FLCs). Plasma cell morphology varies in LCMM, from anaplasia to a degree of maturation. Rarely, signet ring-like morphology is visualized6. In the present case, the patient was a 29-year-old male.
LCM presents with hypogammaglobulinemia and is associated with Bence–Jones proteinuria. FLCA measures the free kappa-to-lambda chain ratio; the normal range is 0.26 – 1.65. If the ratio is < 0.26, the patient has lambda-chain myeloma; patients with a ratio >1.65 have kappa-chain myeloma4. The more common type is lambda-chain myeloma. LCMs have light chain deposition in various organs, including the heart, liver, kidneys, and the integumentary and central nervous systems. Bone involvement and systemic light-chain AL amyloidosis (in 5 — 10% is cases) is more frequent in cases of LCMM6. In this case, blood FLCA revealed a free kappa light chain level of 16,100 mg/L, free lambda chain level of 7.80 mg/L, and a kappa-lambda ratio of 2064.103. Therefore, a final diagnosis of LCM was proposed.
LCM has poor prognosis as compared with IgA or IgG variants. As the disease progresses, the observed complications include lytic bone lesions, anemia, hypercalcemia, pleural effusion, extramedullary plasmacytoma, increased LDH level, elevated serum FLC level, and higher international staging system scores. Some patients can also develop Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal gammopathy, Skin changes (POEMS) syndrome. Renal involvement is more common in LCMM than in other variants of MM due to the circulating FLC, which causes tubular nephropathy. Very rare clinical presentations in LCMM include liver plasmacytoma and epidural plasmacytoid tumor. LCM can progress to secondary plasma cell leukemia with peripheral blood eosinophilia. This progression is thought to be triggered by functional or genetic alterations. LCMM is the third most common type of MM and is associated with poor prognosis. The overall poor survival is linked to skeletal destruction and lack of differentiation5, 9, 10. The patient in this case presented with multiple lytic bone lesions. The biochemical investigations, including serum calcium and serum creatinine, were normal, except for a mild decrease in serum globulin levels, indicating no significant end-organ damage.
When there is clinical suspicion of MM, serum protein electrophoresis, serum immunofixation, and serum FLCA should be evaluated for detection of M protein6. Initial bone marrow studies at the time of diagnosis should incorporate fluorescent in situ hybridization (FISH) to detect translocations t(14;16), t(6;14), t(14;20) t(11;14), and t(4;14). Conventional karyotyping has some value in detecting hypodiploidy and deletion 13; however, FISH is required for risk stratification. Gene expression profiling (GEP) has added prognostic utility. Radiological investigation of the skeleton is essential for assessing the extent of the bone disease. Serum and urine protein electrophoresis is utilized to monitor M-protein level for assessing treatment response. Serum FLCA is utilized to monitor patients with MM who lack a measurable M protein, especially to diagnose LCMM5. In the present case, FNA, bone marrow aspiration, and Tru-Cut biopsy revealed plasma cells in sheets, favoring a plasma cell dyscrasia; however, serum electrophoresis did not show an M spike. FLCA helped to reach the diagnosis. Molecular genetics investigations were not conducted due to non-availability of the facilities.
Conclusion
Absence of M protein in the serum and urine does not rule out a diagnosis of MM. Instead, diagnosis of other types of myelomas, such as LCM and NSM, should be considered in patients with characteristic laboratory or radiological investigations and clinical presentation. FLCA and estimation of free kappa-lambda light-chain ratio helps in reaching a specific diagnosis.
Abbreviations
None.
Acknowledgments
None.
Author’s contributions
All authors equally contributed to this work, read and approved the final manuscript.
Funding
None.
Availability of data and materials
None.
Ethics approval and consent to participate
This study was conducted in accordance with the amended Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
-
Molloy
C.B.,
Peck
R.A.,
Bonny
S.J.,
Jowitt
S.N.,
Denton
J.,
Freemont
A.J.,
An unusual presentation of multiple myeloma: a case report. Journal of Medical Case Reports.
2007;
1
(1)
:
84
.
View Article PubMed Google Scholar -
Sharma
M.,
Shah
S.,
Champaneria
N.,
Goswami
S.,
Multiple myeloma in 33 years old male patient - A case report. International Archives of Integrated Medicine.
2017;
4
(4)
:
75-8
.
-
Bensalah
M.,
Lamrabat
S.,
Lyagoubi
A.,
Aarab
A.,
Bouayadi
O.,
Seddik
R.,
A Rare Case of Non-Secretory Multiple Myeloma: A Case Report and Literature Review. The journal of the International Federation of Clinical Chemistry and Laboratory Medicine.
2019;
30
(1)
:
88-94
.
PubMed Google Scholar -
Singh
T.,
Atlas and Text of Hematology. 4th ed. New Delhi: Avichal publishing company. 2018. p 455-87.
.
-
Rajkumar
S.V.,
Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. American Journal of Hematology.
2020;
95
(5)
:
548-67
.
View Article PubMed Google Scholar -
Rafae
A.,
Malik
M.N.,
Abu Zar
M.,
Durer
S.,
Durer
C.,
An Overview of Light Chain Multiple Myeloma: Clinical Characteristics and Rarities, Management Strategies, and Disease Monitoring. Cureus.
2018;
10
(8)
:
e3148
.
View Article PubMed Google Scholar -
Duncan
N.,
Multiple myeloma clinical features and classification. Clinical Pharmacy.
2012;
4
:
124-6
.
-
Caldas
A.R.,
Brandao
M.,
Marinho
A.,
Non secretory myeloma or Light chain myeloma: A Case Report. Journal of Medical Case Reports.
2011;
2
:
97-100
.
-
Pavan
M.,
Ashwini
K.A.,
Ravi
R.,
Suratkal
L.H.,
Complete remission of lambda light chain myeloma presenting with acute renal failure following treatment with bortezomib and steroids. Indian Journal of Nephrology.
2010;
20
(2)
:
94-6
.
View Article PubMed Google Scholar -
Singh
N.,
Agrawal
N.,
Sekhri
R.,
Mehta
A.,
Kumar
D.,
Vishwakarma
G.,
Light chain myeloma: A brief report from India. Indian Journal of Pathology & Microbiology.
2019;
62
(3)
:
441-4
.
View Article PubMed Google Scholar
Comments
Article Details
Volume & Issue : Vol 9 No 9 (2022)
Page No.: 5272-5277
Published on: 2022-09-30
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 5263 times
- PDF downloaded - 1477 times
- XML downloaded - 0 times
