

Copyrights: Nursyuhana Rason, Ida Shazrina Ismail, Eshaifol Azam Omar, Nor Azlina Khalil, Seoparjoo Azmel Mohd Isa, Sellamuthu Subbana Gounder, Baskar Subramani, Rafeezul Mohamed, 2022. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Background: Different types of immune cells, such as tumor-specific T cells, natural killer (NK) cells and dendritic cells (DCs), are being studied for use in treating metastatic cancer. However, disease progression varies based on the source, type, and mode of administration of immune cells. The aim of the current study is to determine the effect of intravenous administration of human peripheral blood-derived CD8+ T cells and tumor-activated monocyte-derived dendritic cells (moDCs) pulsed CD8+ T cells on a human metastatic breast cancer mouse model.
Methods: A total of nine NOD.SCID gamma (NSG) mice were randomized into three groups to investigate their therapeutic effect. After induction of MDA-MB-231, the NSG mice were monitored daily for well-being and tumor growth. Once tumor size was approximately one millimeter, the CD8+ T cells and tumoractivated moDCs pulsed CD8+ T cells were injected intravenously twice a week for three weeks. A month after the last treatment, all NSG mice were terminated and tumor prognosis was evaluated by performing histological analysis on the mammary fat pads and livers. Interferon (IFN)-g , interleukin (IL)-4, IL-10 and IL-17 levels were evaluated in the serum using luminex cytokine array and T-bet, GATA-3, Foxp3, ROR-gamma t gene expression in the mammary fat pad and the liver were measured using quantitative real-time polymerase chain reaction (qRT-PCR).
Results: Tumor-bearing NSG mice treated with CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells significantly reduced tumor necrosis in the liver but not in the mammary fat pad compared to untreated tumorbearing NSG mice. No difference was seen in the secretion levels of IFN-g , IL-4, IL-10, and IL-17 in the serum of untreated and treated tumor-bearing NSG mice before and after treatment. Mammary fat pad and liver isolated from the tumor-bearing NSG mice treated with CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells showed increased T-bet gene expression but reduced GATA-3, Foxp3, and RORgamma t gene expression.
Conclusion: The current study indicated that tumoractivated moDCs pulsed CD8+ T cells treated mice showed better outcomes in reducing tumor necrosis compared to CD8+ T cells, which is supported by increased anti-tumor related gene expression and reduced pro-tumor related gene expression in the mammary fat pads and livers of tumor-bearing NSG mice.
Introduction
It was estimated that 2.3 million new breast cancer cases were detected among women in 2020, which represented 11.7% of new cancer cases, with an estimated 684,996 deaths globally1. Additionally, breast cancer represents one in four of all cancers and causes one in six deaths in women1. The occurrence of breast cancer is anticipated to increase more than 46% based on the GLOBOCAN Cancer Tomorrow prediction tool2. In 2018, the total number of the new cases among Malaysian women was 7593 (32.7%), causing 2894 deaths3. Generally, 30–40% of Malaysian women were diagnosed at stage 3 and 4 breast cancer compared to other countries in Asia4, 5. Treatment for a metastatic patient is a very complex issue, with the patient often developing resistance to available chemotherapeutic agents. After exhausting the first or second line treatments, there are usually limited chemotherapeutic options available, or patients may not be suitable for further chemotherapy due to the toxicities of the previous treatments. At this stage, further treatment may significantly increase morbidities and any further survival or symptomatic benefits may be minimal. Therefore, the establishment of new therapeutic approaches to treat metastatic breast cancer patients are urgently needed.
Tumor-specific immunotherapy is a therapeutic approach recently introduced to treat metastatic breast cancer patients. Systematic activation and enhancement of the host immune system facilitates the induction of tumor-specific anti-tumor immune responses, which can easily recognize and eradicate the tumor cells as well as induce the immunological memory to control tumor relapse6. This theoretical clinical approach could be an appropriate treatment regimen for a heterogeneous population of metastasized cancer. Current evidence suggests that antigen-specific cytotoxic T lymphocyte (CTL) cells play a crucial role in development of immunotherapy7. Additionally, dendritic cells (DCs) are critical in the development of antigen-specific effector cells and generating therapeutic immunity against cancers8. One role of DCs is to induce tumor-specific CTLs that function in recognition and elimination of antigen-specific cancer cells9. Notably, DCs can carry more than one antigen and can therefore generate more than one antigen-specific CTL clone. This could facilitate the eradication of the heterogeneous cancer cell population, including the cancer stem cell (CSCs)10. Furthermore, with this widely expanding knowledge, numerous in vitro and pre-clinical studies have successfully demonstrated that autologous immune enhancement therapy (AIET) is feasible and tolerable, with promising results for various metastatic cancers11.
AIET offers a new therapeutic strategy to overcome the limitations of current therapeutic regimens and to improve the treatment outcomes in patients with advanced breast cancer. Both innate (DCs) and adaptive (T cells) immune cells play a crucial role against cancer development in a process called “anti-cancer or immune response”. This AIET is designed for systematic activation and production of an antigen-specific immune response that can recognize tumor antigens expressed on the surface of cancer cells. Ultimately, the objective of active immunotherapy is to elicit antigen-specific T cell responses. These antigen-specific immune responses are mostly dependent on the presence of DCs in lymphoid tissues and peripheral blood. These DCs will uptake the tumor-specific antigens and process them into small peptides that are then coupled with either major histocompatibility complex ((MHC)-I or MHC-II) molecule expressed on the DCs surface12. These antigen-presenting cells (APCs) migrate into the lymphoid organ and are bound with Th1 (CD4+ T cell) cells through the MHC-II molecule, stimulating the secretion of the cytokine milieu, which includes IL-2 and IFN-γ, which is essential for activation and proliferation of the antigen-specific CTLs12. Alternatively, the MHC-I molecules on APCs are bound to CD8+ T cells and generate antigen-specific CTLs in the presence of Th1 cytokines12. Additionally, MHC-II molecules on APCs can bind with Th2 cells to stimulate the secretion of IL-4 and IL-10, as well as interacting with B cells to stimulate antibody production12.
Most metastatic breast cancer patients show defects in anti-tumor immunity, which includes decreased DC maturation, impaired natural killer (NK) response, CTL cell maturation, and cytotoxic function, while T regulatory (Treg) cell infiltration is increased13. Hypothetically, poor host immunity may facilitate breast cancer cells escaping anti-tumor immunity, leading them to eventually metastasize. This clearly indicates that metastatic breast cancer is strongly linked with anti-tumor immune efficiency, particularly DCs, CTLs, and NK cells. This, in line with specific reactivation of anti-tumor immunity anticipate potential clinical outcome in patients with advanced breast cancer. The present study aimed to evaluate the synergistic effect of ex vivo prepared peripheral blood-derived CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells in a mouse model bearing metastatic breast cancer.
Methods
Ethics statement
The leukapheresis procedure was carried out after obtaining a letter of approval from Universiti Sains Malaysia (USM) human ethical research committee (JEPeM code: USM/JEPeM/17120677). The animal study was endorsed by the USM Institutional Animal Care and Use Committee (IACUC) (USM/Animal Ethics Approval / 2015 / (96)/ (628)). All animals were kept and preserved based on the guidelines stated by IACUC.
Leukapheresis
Leukapheresis was carried out through peripheral vein entree using the Spectra Optia system (Terumo BCT, Lakewood, Colorado, USA) based on the CMNC program (software version 11.3) as previously described14. The anticoagulant utilized in the collection bag was acid-citrate-dextrose formula A (ACD-A) (Terumo BCT, Lakewood, Colorado, USA) without heparin. The flow rate was positioned at 0.8 ml/min/LTBV with a ratio of 1:12 between anticoagulation and inlet. The inlet flow rate during the procedure ranged from 40 to 70 ml/min while the anticoagulant flow rate ranged from 0.7 to 1.1 ml/min/LTBV. The flow rate for the collection was fixed at one ml/min. Approximately 120 ml of mononuclear cells (MNCs) was obtained from the leukapheresis procedure.
CD8+ T cells isolation
Human CD8+ T cells were separated from freshly isolated MNCs by negative selection using the human CD8+ T cell isolation kit as previously described15 and these were cultured in appropriate medium in the good manufacturing practice (GMP)-certified laboratory owned by Nichi-Asia Life Science Sdn Bhd, Malaysia. Briefly, 2 x 108 cells were mixed into 400 µL of PBS, then 100 μL of non-CD8+ T cell biotin-antibody cocktail was added to the mixture and incubated at 4 °C for 5 minutes. Next, 300 μL of phosphate-buffer saline (PBS) and 200 μL of anti-biotin microbeads were added in the cell mixture and incubated at 4 °C for 10 minutes. The CD8+ T cells were separated by filling cells into the LS separation column attached to the magnetic cell separator. The flow-through consisting of unlabeled cells, which was enriched with CD8+ T cells, was collected, while labelled cells (non-CD8+ T cells) were trapped in the column due to the magnetic beads bound to their surface antigens. After separation, the suspension was removed and centrifuged at 300 x g for 10 minutes. The cell pellet was mixed with one ml complete RPMI medium and the cells were counted.
Human moDCs production
Human moDCs were produced by culturing freshly isolated MNCs in culture plates for 2 hours in the GMP-certified laboratory owned by Nichi-Asia Life Science Sdn Bhd, Malaysia. Non-adherent cells were discarded while adherent cells (monocytes) were cultured in complete RPMI 1640 which was composed of 10% FBS, 1 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin, together with human granulocyte macrophage colony-stimulating factor (GM-CSF) at 50 ng/ml (Peptrotech, Rocky Hill, NJ, USA) and 10 ng/ml recombinant human IL-4 (Peprotech, Rocky Hill, NJ, USA) for 7 days in the 5% CO2 incubator at 37 °C. The medium was changed every two days with fresh complete RPMI medium together with GM-CSF and IL-4. Immature moDCs were successfully produced after 7 days.
CD8+ T cells and moDC phenotyping
Isolated CD8+ T cells and generated moDCs were characterized by staining CD8+ T cells for CD3 and CD8, and moDCs for CD11c and CD83 surface expression. The phenotyping and analyses were carried out using BD FACSCanto II (Becton Dickinson, Franklin Lakes, New Jersey, USA) with FlowJo 7.5.3 software.
Culturing MDA-MB231 and preparation of cells lysate
MDA-MB-231 originating from human breast adenocarcinoma cell lines was obtained from the American Type Culture Collection (Manassas, Virginia, USA) and maintained in the Dulbecco's Modified Eagle's medium (DMEM) (Thermo Fisher Scientific, Waltham, Massachusetts, USA) containing 5% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, Massachusetts, USA), 100 U/ml penicillin, 100 µg/ml streptomycin (Thermo Fisher Scientific, Waltham, Massachusetts, USA), 600 µg/ml L-glutamine (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and 6/500 ml 100X non-essential amino acids (Thermo Fisher Scientific, Waltham, Massachusetts, USA), and kept in the 5% CO2 incubator at 37 °C in the GMP-certified laboratory (Nichi-Asia life science Sdn Bhd, Malaysia). MDA-MB-231 cells were accumulated at the exponential phase of growth before injection in the mammary fat pads of mice.
Antigen pulsing of DCs
Immature DCs were collected on day 7, and 10 x 106 cells were plated into a T25 flask. Mature DCs were induced with whole tumor lysate from MDA-MB-231 cells. Briefly, tumor cells were collected at 80% confluence by depleting using 2 ml 0.25% trypsin-EDTA, incubated in the 5% CO2 incubator at 37 °C for 5 min, and washed in 5 ml of PBS. The trypsinased cells were centrifuged at 300 x g for 5 minutes. The cell pellets were lysed using the RIPA Lysis Buffer System. The BCA Protein Assay Kit was used to determine the total protein concentration. The tumor lysates were stored in aliquots at -80 °C until further use. Tumor lysates were mixed with DCs in the culture flask at a ratio of one DC to five tumor cell equivalents for 24 hours.
Tumor-activated moDC pulsed CD8+ T cells
Mature moDCs and T cells were co-cultured in a T25 flask at a ratio of 1∶20 (DCs:T), as this ratio was indicated to produce the most desired T cell proliferation. This high ratio was expected due to the relative infrequency of DCs bearing in vivo-captured antigen. Co-cultures were preserved for 48 hours for further experiments.
Animals and treatment
Eight weeks old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were acquired from Jackson Laboratory (Bar Harbor, Maine, USA). After one-week of acclimation to the animal research facility, Advanced Medical and Dental Institute, USM, NSG mice were subjected to tumor inoculation. MDA-MB-231 cells were suspended into 200 μl of PBS at a concentration of 5 x 106 cells. Before tumor inoculation, mice were anesthetized with ketamine-xylazine-acepromazine (80 – 120 mg/kg) by intra-peritoneal injection. A small incision was made over the right or left auxiliary mammary fat pad and the tumor cells were injected using an insulin syringe with a 27 to 30-gauge needle. The incision was closed with a simple suture. Mice were monitored daily for well-being (physical behavior, food and drink update, and animal body weight) and tumor development (tumor volume and size). Tumor size was measured twice a week using caliper measurement and the volume were calculated as D x d2 x 0.52, where D is the long diameter and d is the perpendicular short diameter. After tumor size reached approximately 0.2 cm, NSG mice were injected via tail vein with 20 x 106 CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells twice a week for 3 weeks. Treated NSG mice were terminated four weeks after immune cell treatment. Blood samples were collected from the submandibular vein of NSG mice at the experiment initiation and before termination. After treated NSG mice were terminated, primary mammary fat pad tumor tissue and lung were harvested. Harvested tumor organs were been subjected to pathophysiological examination.
Histology
Formalin-fixed mammary fat pad and liver were fixed with paraffin, cut at 5 μm, and stained with haematoxylin and eosin using the established procedure. Briefly, the sectioned tissue on the glass slides were subjected to deparaffinization using xylene for 2 minutes, rehydration using various percentages of graded ethanol (100%, 90%, 80%, and 70%) each for 2 minutes, left under tap water flow for 2 minutes, stained with Harris’ haematoxylin (Sigma Aldrich, St. Louis, Missouri, USA) for 11 minutes, 0.5% acid alcohol for 2 seconds, tap water for 2 minutes, 0.2% ammonia water (Merck, Kenilworth, New Jersey, USA) for 20 seconds, and tap water for 2 minutes. Then, the sample was counter-stained with eosin (Sigma Aldrich, St. Louis, Missouri, USA) for 3 minutes. The dehydration process was started by using graded ethanol (70%, 80%, 90%, and 100%) for 10 seconds followed by clearing using xylene for 2 minutes. The slides were then mounted using DPX mountant (Sigma Aldrich, St. Louis, Missouri, USA) and the staining was visualized using an Olympus CX31 light microscope (Olympus, Shinjuku City, Tokyo, Japan).
Measurement of cytokines in mouse serum
Measurement of IFN-γ, IL-4, IL-10, and IL-17 in mouse serum from blood that was collected from the submandibular veins at the initiation, and before the end, of the experiments. Bloods were centrifuged at 2000 x g for 10 minutes to isolate serum from other blood components. IFN-γ, IL-4, IL-10, and IL-17 secretion was evaluated by using mouse magnetic luminex assay (R&D Systems, McKinley Place, Minneapolis, Minnesota, USA) and analyzed on the Luminex® 100 (Luminex Corp., Austin, Texas, USA).
Total RNA extraction and cDNA synthesis
The collected mammary fat pad and liver were cut removed and kept in RNAlater® solution (Ambion™). Approximately 30 mg of the mammary fat pad and the liver were transferred in a lysing matrix D tube (MP Biomedicals, Irvine, CA, USA). TRIzol was added into the tube and FASTPrep®-24 (MP Biomedicals, Irvine, CA, USA) was used to homogenize the tissues. The homogenates were transferred into a new tube and 200 μl of chloroform was added. The cap of the sample tubes was closed tightly, and tubes were vortexed firmly for 15 seconds. Then, the sample tubes were incubated for 2 to 3 minutes at room temperature. Sample tubes were centrifuged at 12,000 x g for 15 minutes at 4 °C to produce four layers: a colorless upper aqueous phase, interphase, phenol-chloroform phase, and lower red. The upper aqueous phase composed of RNA was relocated carefully into the new assigned tube. Isopropyl alcohol (0.5 ml) was added into the tube containing the aqueous phase and the tube was placed at room temperature for 10 minutes. Following incubation, that tube was centrifuged at 12,000 x g for 10 minutes at 4 °C. The supernatants were discarded, and the RNA pellets were mixed with 1 ml 75% ethanol followed by spinning at 7,500 x g for 5 minutes at 4 °C. The RNA pellets were air-dried for 5–10 minutes and were eluted into 30 μl of RNAase free water. Subsequently, the tetro cDNA synthesis kit (Bioline, London, UK) was utilized to reverse-transcribe 2 μg of total RNA. Briefly, the reaction mixture tube containing total RNA, ribosafe RNA inhibitor, oligo (dT) 18, 5x RT buffer 10 mM dNTP, tetro reverse transcriptase, and DEPC-treated water, totaling 20 μl, was prepared. The sample tubes were vortexed firmly and incubated at 45 °C for 30 minutes. The reactions were terminated by incubating at 85 °C for 5 minutes and placed on ice.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
A SensiFAST SYBR Hi-Rox Kit (Bioline, London, UK) was used to quantify relative mRNA expression levels on a StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The reaction mixtures containing 10 μl of 2x SensiFAST SYBR® Hi-ROX Mix, 0.8 μl of 10 μM primer pair mixture, 7.4 μL H2O and 1 μl of cDNA were prepared and subsequent qRT-PCR was carried out as follows: a denaturing step at 95 °C for 2 min, then 95 °C for 10 s (40 cycles) and finally 60 °C for 30 s. The sequences of the primers for target genes were T-bet: 5’-CCTGGACCCAACTGTCAACT-3’ (F) 5’-AACTGTGTTCCCGAGGTGTC-3’ (R), RORγt: 5’TGCAAGACTCATCGACAAGG-3’(F) 5’-AGGGGATTCAACATCAGTGC-3’ (R), Foxp3: 5’-CAACCTAGCCCCAAGATGAA-3’ (F) 5’-CCAGATGTTGTGGGTGAGTG-3’ (R), and GATA-3: 5’-CCGAAACCGGAAGATGTCTA-3’ (F) 5’-AGGGCTCTGCCTCTCTAACC-3’ (R). ΔΔCt method was used to calculate the relative mRNA expression.
Statistical analysis
GraphPad Prism Version 8.0 software was used to carried out statistical analysis. Values of the data were presented as mean ± standard deviation (SD) from three mice per treatment group and were analyzed using one-way analysis of variance (ANOVA) followed by the Bonferroni post-hoc test. The expressions of IFN-γ, IL-4, IL-10, and IL-17 in the serum of blood were analyzed by two-way ANOVA followed by Bonferroni post hoc test.





Results
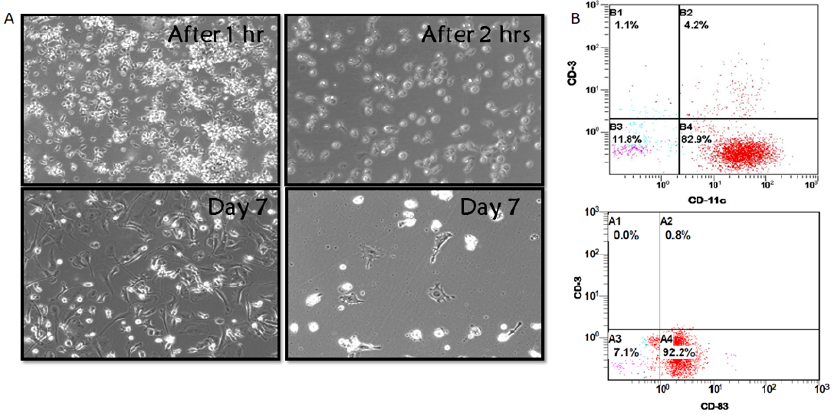
Characterization of expanded CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells
We examined expanded CD8+ T cells by performing basic phenotyping for CD3+CD8+ cells using flow cytometry analysis. Figure 1A shows that the percentage of CD3+CD8+ T cells was 74.6%. The CD8+ T cell growth from two different donors showed consistent shape as indicated by microscopy (Figure 1 B). After 1 and 2 hours of culture, the microscopy showed round-shaped and non-adherent cell characteristics (Figure 2 B). However, the generated moDCs appeared as single cells or loosely adherent aggregates (Figure 2 B). The generated moDCs were comprised of 82.9% CD3-CD11c+ population and CD3-CD83+ population. MDA-MB-231 cell lysate-activated moDCs pulsed CD8+ T cells at a ratio of 1 in 20 stimulated CD8+ T cell proliferation (Figure 3).
Tumor-activated moDCs pulsed CD8+ T cells reduced tumor necrosis
After approximately 2 months, tumor-bearing NSG mice developed primary metastatic breast cancer, as the anatomy of the mammary fat pad and the liver following haematoxylin and eosin staining showed the presence of numerous macroscopic metastatic foci in untreated and treated tumor-bearing NSG mice (Figure 4). The shape and size of the mammary fat pad and the liver did not show any clear differences between untreated and treated tumor-bearing NSG mice, with the size of both organs being 1.6 – 2.0 mm in all cases. No significant reduction of tumor necrosis in the mammary fat pad was seen between tumor-bearing NSG mice treated with CD8+ and tumor-activated moDCs pulsed CD8+ T cells and untreated tumor-bearing NSG mice (Figure 4 A). However, tumor-bearing NSG mice treated with CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells significantly reduced tumor necrosis in the liver compared to untreated tumor-bearing NSG mice (p < 0.05 and p < 0.01, respectively) (Figure 4 B).
No changes in the secretion of IFN-γ, IL-4, IL-10 and IL-17
We assessed the expression of IFN-γ, IL-4, IL-10, and IL-17 in the serum of blood collected from the submandibular vein of untreated and treated tumor-bearing NSG mice to investigate their potential association with tumor progression. Our results indicated that IFN-γ, IL-4, IL-10, and IL-17 secretion in the blood of untreated and treated tumor-bearing NSG mice did not show any significant differences at experiment initiation and after termination.
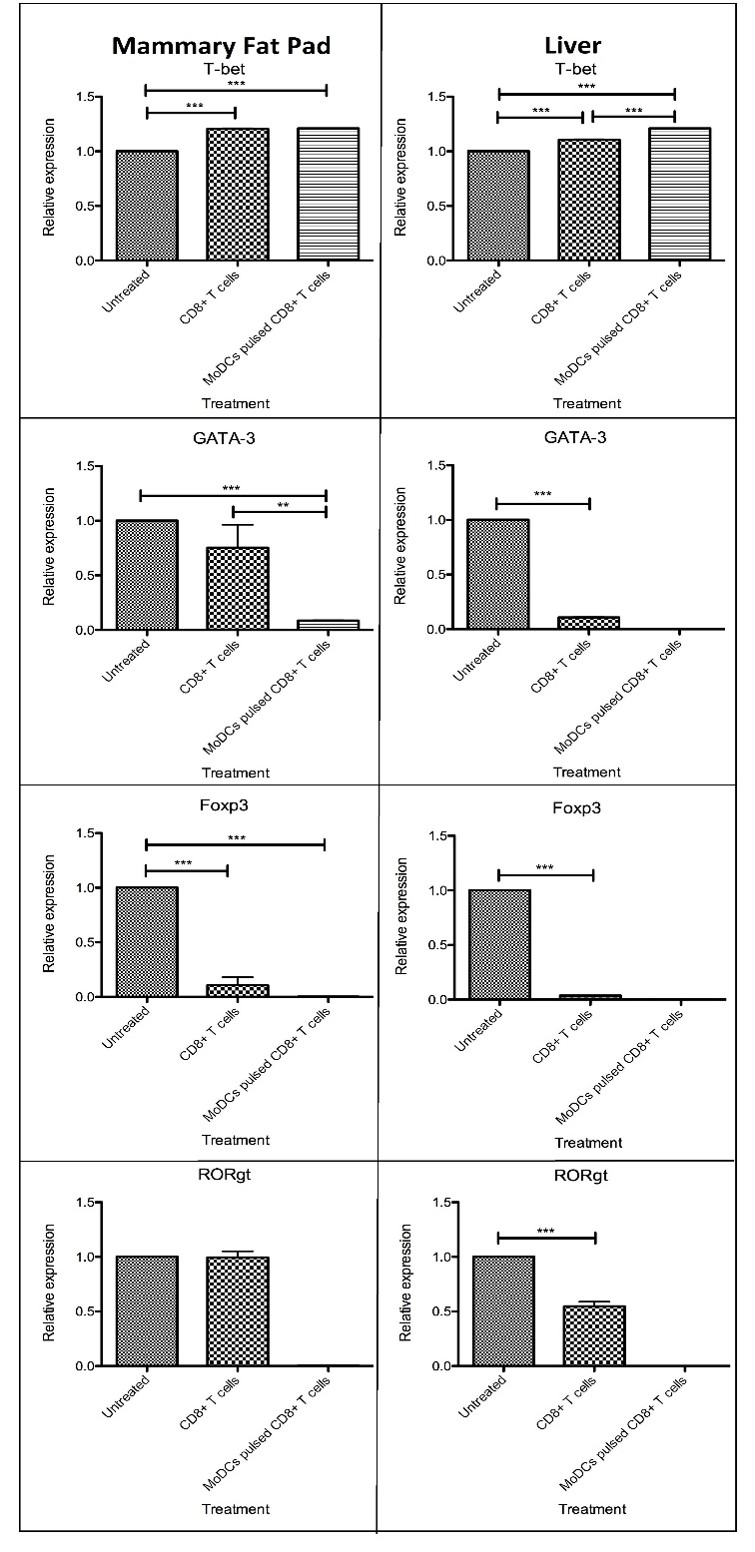
CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells increased anti-tumor related gene, but reduced pro-tumor related gene, expression
We also evaluated the expression of Th1, Th2, Treg, and Th17 related transcription factor (TF) genes in the mammary fat pads and livers of untreated and treated tumor-bearing NSG mice to associate these with tumor necrosis. The mRNA level of Th1-specific TF, T-bet, was increased in the mammary fat pad and the liver of tumor-bearing NSG mice treated with CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells (p < 0.001) compared to untreated tumor-bearing NSG mice. Interestingly, tumor-bearing NSG mice treated with CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells had reduced expression of Th2-specific TF, GATA-3, and Treg-specific TF, Foxp3, in the mammary fat pad and the liver (p < 0.001) compared to untreated tumor-bearing NSG mice. However, only the livers of tumor-bearing NSG mice treated with CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells showed a reduction of Th17-specific TF, RORγt (p < 0.001) compared to untreated tumor-bearing NSG mice.
Discussion
The correlation between immune system responses and tumor development is being intensively investigated in metastatic breast cancer. Tumor antigen-specific CD8+ T cell-based immunotherapy has appeared as one of the most potent therapies for various types of cancer16, 17, 18. However, this treatment regime is hindered by low T cell survival following their introduction in vivo19. The current study was performed to evaluate the effect of CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells injection in a metastatic breast cancer mouse model. The foundation of this treatment is the induction of an anti-tumor response by CTLs following the engagement of tumor antigens with DC receptors in the patient20. Engagement of both DCs and tumor-associated antigens (TAAs) are crucial as DCs are regarded as the optimal APCs for recognizing and processing a wide array of TAAs, such as peptides, heat shock proteins, lysates and apoptotic bodies, to induce tumor based immune responses20.
Initially, isolated CD8+ T cells were isolated from MNCs using a CD3 gate to discriminate T cell lymphocytes, with subsequent analysis of CD8 expression within the CD3 gate using flow cytometry. 74.6% of the population was CD3+CD8+, which was acceptable percentage. Also, moDCs were successfully generated from MNCs using GM-CSF and IL-4. These moDCs displayed single cells or loosely adherent aggregates and expressed CD11c and CD83, which are both DC-related markers. CD11c is also recognized as integrin alpha X and is most frequently utilized as a characterizing marker for DCs21. CD83 has a critical role as a co-stimulatory signal for the activation of naïve and memory T cells22. Therefore, CD8+ T cells and functional moDCs were successfully isolated and induced from MNCs respectively, and both types of these cells were utilized in subsequent experiments.
These moDCs were activated with tumor lysates originating from MDA-MB-231 cell lines before co-culturing with CD8+ T cells in a ratio of 1:20. Then, CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells were injected via the tail vein of tumor-bearing NSG mice. NOD.SCID gamma (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) was used in this study as NSG mice are a widely accepted xenograft model for studying advanced metastatic breast cancer, and they displayed the generation of metastasis originating from the mammary gland following induction with human cancer cells23. NSG mice are prone to metastasis generation due to complete depletion of B cells, NK cells, T cells, DCs, macrophages, and the complement system, and complete engraftment and cell growth has been reported upon injection of human breast cancer cell lines into the NSG mice23. Therefore, NSG mice are the most utilized model for replicating the human environment for studying the underlying mechanism for the eradication of metastatic breast cancer by cancer-specific adaptive immune therapy23.
Our findings showed that tumor-activated moDCs pulsed CD8+ T cells decreased tumor necrosis in both the mammary fat pad and liver whereas CD8+ T cells only attenuated tumor necrosis in the liver. DCs have an important role in eliciting T cell anti-tumor responses24, 25. DCs originating in the tumor-draining lymph nodes are involved in engaging tumor antigens and presenting them to naive CTLs26, 27. For example, CD8α+ DCs residing inside lymph nodes were specifically responsible for the engagement of exogenous antigens, processing and presenting peptides on their surface to naive CD8+ T cells using MHC-I28, whereas migratory CD8α− DCs are involved in the presentation of peptides to CD4+ T cells using MHC-II29. DCs play a pivotal role in regulating T cell anti-tumor responses, and the current study has shown that tumor-activated moDCs pulsed CD8+ T cells produce a better outcome for reducing tumor metastasis. Another study has shown that CD8α+ DCs were required for eliciting CTL anti-tumor responses in transgenic mice depleted of transcription factor Batf330. These mice displayed a deficiency of CD8α+ DCs in their spleen and lymph nodes, while other subsets of DCs remained functional and intact. In the current study, the ineffectiveness of the CTL treatment in reducing tumor necrosis in the mammary fat pad may be due to a failure to process and present tumor antigens on the MHC-II complex rather than there being a deficiency in cross-presentation26.
The adoptive transfer of several types of lymphocytes for managing breast cancer has been investigated in numerous studies. The transplant of allogeneic stem cells in combination with high-dose chemotherapy produced robust results but also elicited significant safety concerns, whereas adoptive cell therapy produced better outcomes but displayed a lack of efficacy31, 32, 33, 34. The adoptive transfer of tumor-infiltrating lymphocytes (TILs) in breast cancer patients revealed a clear correlation between eliciting stromal TIL populations and a better prognosis in triple-negative breast cancer35, 36, 37, 38. DCs are utilized for breast cancer immunotherapy because they can activate CD8+ T cells and CD4+ T cells, eliciting memory T cells which generate an additional cytotoxic effect when combating tumors39. Autologous DCs conjugated with tumor cells via recognition with specific receptors or activated by either tumor antigens or tumor lysates have been used to induce T cell anti-tumor responses40, 41, 42. In contrast to the findings demonstrated in TILs and T cell receptor therapies, large numbers of DCs can be generated from the moDCs of peripheral blood and bone marrow precursors via the apheresis procedure43. Currently, almost 20 clinical studies are ongoing at different phases to evaluate the effectiveness of DC vaccinations in breast cancer patients with all major pathologies, with most studies constructed to activate DCs with selected, well-known tumor antigens44. Although adoptive cell therapy with DCs did not show any significant outcomes during clinical trial phases44, the results of the current study justify the role of DCs in stimulating a T cell response, providing durable memory for the treatment of breast cancer. Additionally, DCs are safe to use and have well-established methods for manufacturing in a GMP laboratory, giving them an advantage for utilization, alone or in combination with other T cell therapies, for breast cancer treatment44.
Subsequently, we also evaluated the secretion of selected cytokines in the serum of tumor-bearing NSG mice treated with CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells. The augmentation of breast cancer not only relies on its malignant cells, but also involves the interplay between secreted growth factors and cytokines that are generated by a wide array of cells such as malignant, stromal, immune, and endothelial cells populating the tumor microenvironment45. Th1 cells mainly produce IL-2, TNF-α and IFN-γ, which elicit macrophage cytolytic and anti-tumor activities46. Th2 cells secrete IL-5, IL-6, IL-10, IL-13, and IL-4, which stimulate T cell anergy and induce the activity of tumor-associated macrophages (TAMs)46. A recent study indicated that a Th17 cell secreted cytokine, IL-17A, promoted tumor progression by altering the non-metastatic tumor cell gene expression profile47, resulting in a pro-tumor effect. Additionally, increased IL-17A expression corresponded with a poor prognosis in patients with invasive ductal carcinoma (IDC) of the breast47. Treg cells stimulated breast cancer growth by hindering the activity of anti-tumor CTL and Th1 cells48. High Foxp3 expression also indicated that a high infiltration level of Treg cells was closely correlated with the size, vascularity, and invasion of the tumor48. The current study did not show any significant differences in the secretion of IFN-γ, IL-10, IL-4, or IL-17 in the serum of all groups of tumor-bearing NSG mice before and after injection of CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells. The expression of Th1-specific TF T-bet was increased in the mammary fat pad and liver of tumor-bearing NSG mice treated with CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells. Interestingly, CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells had reduced expression of Th2-specific TF GATA-3 and Treg-specific TF Foxp3 in the mammary fat pads and the livers of tumor-bearing NSG mice. However, only the liver of tumor-bearing NSG mice treated with CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells showed a reduction of Th17-specific TF RORγt. The reduction of tumor metastasis in tumor-bearing NSG mice treated with CD8+ T cells and tumor-activated moDCs pulsed CD8+ T cells, especially in the liver, was associated with the increase of anti-tumor related Th1 cells and reduced pro-tumor associated Th2, Treg, and Th17 cells.
As an alternative to the application of tumor lysate pulsed DCs for eliciting a CD8+ T cell anti-tumor response, total RNA extracted from tumor biopsy and pulsed into DCs has produced encouraging results for inducing effective Th1 and CD8+ T cell responses against hepatoma cells49. Additionally, a study by Sumramsub et al. demonstrated that total RNA extracted from breast CSCs-pulsed DCs stimulated anti-tumor cells, such as CD8+ T cells, Th1 cells, and NK cells, to induce breast cancer apoptosis with greater killing capability than DCs pulsed with total RNA extracted from whole cancer cells, which are composed of a combination of CSCs and non-CSCs50. Moreover, the utilization of RNA hinders the reaction of the immunosuppressive factors derived from the tumor protein lysate. Therefore, DCs pulsed with RNA may produce more potent anti-tumor effects50. RNA-based vaccines are successful due to their simplicity as the RNA encodes the specific genes of interest, is low cost as mRNA can be easily and cheaply extracted at higher quantities, and safe due to the shorter half-life of RNA in cells and with no risk of incorporation into the genome of the transfected cell as the RNA deteriorates upon translation of the encoded protein51. Additionally, the injected RNA molecules are translated into proteins in the cytoplasm of the host cells without the occurrence of transcription in the nucleus, as is required for DNA a vaccine, therefore separating them from host transcription factors51.
Conclusions
The current study showed that tumor-activated moDCs pulsed CD8+ T cells produced a better outcome in reducing tumor necrosis compared to CD8+ T cells, which is supported by increased expression of anti-tumor related genes, and reduced expression of pro-tumor related genes, in the mammary fat pads and the livers of tumor-bearing NSG mice. However, tumor-activated moDCs pulsed CD8+ T cells therapy for breast cancer requires further study in other humanized breast cancer mouse models and could be studied in combination with other cell therapies, chemotherapy, or radiotherapy to enhance their effectiveness against metastatic breast cancer.
Abbreviations
AIET: Autologous immune enhancement therapy; ANOVA: one-way analysis of variance; APCs: Antigen presenting cells; CSCs: Cancer stem cells; CTL: Cytotoxic T lymphocytes; DCs: Dendritic cells; IFN γ: Interferon- γ; GM-CSF: Granulocyte macrophage-colony stimulating factor; GMP: Good manufacturing practice; IL: Interleukin; MHC: Major histocompatibility complex; MoDCs: Monocyte-derived dendritic cells; NK cells: Natural killer cells; NSG: NOD.SCID gamma; qRT-PCR: quantitative Real Time-Polymerase Chain Reaction; SD: Standard deviation; TAA: Tumor-associated antigen; TF: transcription factor; TILs: Tumor-infiltrating lymphocytes; Treg: T regulatory
Acknowledgments
We acknowledge the top management of Nichi Asia Life Science Sdn. Bhd. for allowing us to use their GMP-certified laboratory for preparation of cell products and staffs at transfusion unit, Advanced Medical and Dental Institute for assisting us in carried out leukapheresis.
Author’s contributions
NR carried out the animal study and post animal analysis, ISI and EAO involved in flow cytometry analysis and contributing ideas in drafting the research proposal, NAK assists NR in performing animal study, SAMI evaluates tumor necrosis, SSG and BS involve in preparation of DCs and CD8+ T cells and also involved in designing the whole experiment, RM is the principal investigator for the grant that fund this research, designing the whole experiment, help NR in carried out animal study and wrote the manuscripts. All read and approved the manuscript.
Funding
This study was supported by R&D fund (Project No: 02-01-05-SF0834) from Ministry of Science, Technology and Innovation, Malaysia.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
-
Sung
H.,
Ferlay
J.,
Siegel
R.L.,
Laversanne
M.,
Soerjomataram
I.,
Jemal
A.,
Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a Cancer Journal for Clinicians.
2021;
71
(3)
:
209-49
.
View Article PubMed Google Scholar -
Heer
E.,
Harper
A.,
Escandor
N.,
Sung
H.,
McCormack
V.,
Fidler-Benaoudia
M.M.,
Global burden and trends in premenopausal and postmenopausal breast cancer: a population-based study. The Lancet. Global Health.
2020;
8
(8)
:
e1027-37
.
View Article PubMed Google Scholar -
Bray
F.,
Ferlay
J.,
Soerjomataram
I.,
Siegel
R.L.,
Torre
L.A.,
Jemal
A.,
Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a Cancer Journal for Clinicians.
2018;
68
(6)
:
394-424
.
View Article PubMed Google Scholar -
Abdullah
N.N.,
Aziz
N.A.,
Rampal
S.,
Al-Sadat
N.,
Mammography screening uptake among hospital personnel in Kuala Lumpur tertiary hospital. Asian Pacific Journal of Cancer Prevention.
2011;
12
(10)
:
2643-7
.
PubMed Google Scholar -
Dahlui
M.,
Ramli
S.,
Bulgiba
A.M.,
Breast cancer prevention and control programs in Malaysia. Asian Pacific Journal of Cancer Prevention.
2011;
12
(6)
:
1631-4
.
PubMed Google Scholar -
Palucka
K.,
Banchereau
J.,
Cancer immunotherapy via dendritic cells. Nature Reviews. Cancer.
2012;
12
(4)
:
265-77
.
View Article PubMed Google Scholar -
Farhood
B.,
Najafi
M.,
Mortezaee
K.,
CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. Journal of Cellular Physiology.
2019;
234
(6)
:
8509-21
.
View Article PubMed Google Scholar -
Fu
C.,
Jiang
A.,
Dendritic cells and CD8 T cell immunity in tumor microenvironment. Frontiers in Immunology.
2018;
9
:
3059
.
View Article PubMed Google Scholar -
Harimoto
H.,
Shimizu
M.,
Nakagawa
Y.,
Nakatsuka
K.,
Wakabayashi
A.,
Sakamoto
C.,
Inactivation of tumor-specific CD8+ CTLs by tumor-infiltrating tolerogenic dendritic cells. Immunology and Cell Biology.
2013;
91
(9)
:
545-55
.
View Article PubMed Google Scholar -
Palucka
K.,
Q&A: Evidence presenter. Interview by Marian Turner. Nature.
2013;
504
(7480)
:
9
.
View Article PubMed Google Scholar -
Farkona
S.,
Diamandis
E.P.,
Blasutig
I.M.,
Cancer immunotherapy: the beginning of the end of cancer?. BMC Medicine.
2016;
14
(1)
:
73
.
View Article PubMed Google Scholar -
Hossain
M.K.,
Wall
K.A.,
Use of dendritic cell receptors as targets for enhancing anti-cancer immune responses. Cancers (Basel).
2019;
11
(3)
:
418
.
View Article PubMed Google Scholar -
Gatti-Mays
M.E.,
Balko
J.M.,
Gameiro
S.R.,
Bear
H.D.,
Prabhakaran
S.,
Fukui
J.,
If we build it they will come: targeting the immune response to breast cancer. NPJ Breast Cancer.
2019;
5
(1)
:
37
.
View Article PubMed Google Scholar -
Wölfl
M.,
Greenberg
P.D.,
Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nature Protocols.
2014;
9
(4)
:
950-66
.
View Article PubMed Google Scholar -
Cates
N.C.,
Oakley
D.J.,
Onwuemene
O.A.,
Therapeutic white blood cell and platelet depletions using the spectra OPTIA system continuous mononuclear cell protocol. Journal of Clinical Apheresis.
2018;
33
(5)
:
580-5
.
View Article PubMed Google Scholar -
Restifo
N.P.,
Dudley
M.E.,
Rosenberg
S.A.,
Adoptive immunotherapy for cancer: harnessing the T cell response. Nature Reviews. Immunology.
2012;
12
(4)
:
269-81
.
View Article PubMed Google Scholar -
Rosenberg
S.A.,
Yang
J.C.,
Sherry
R.M.,
Kammula
U.S.,
Hughes
M.S.,
Phan
G.Q.,
Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clinical Cancer Research.
2011;
17
(13)
:
4550-7
.
View Article PubMed Google Scholar -
Maus
M.V.,
Fraietta
J.A.,
Levine
B.L.,
Kalos
M.,
Zhao
Y.,
June
C.H.,
Adoptive immunotherapy for cancer or viruses. Annual Review of Immunology.
2014;
32
(1)
:
189-225
.
View Article PubMed Google Scholar -
Saito
H.,
Okita
K.,
Chang
A.E.,
Ito
F.,
Adoptive transfer of CD8+ T cells generated from induced pluripotent stem cells triggers regressions of large tumors along with immunological memory. Cancer Research.
2016;
76
(12)
:
3473-83
.
View Article PubMed Google Scholar -
Pham
V.,
Nguyen
S.,
Mai
T.,
Phan
N.K.,
Van Pham
P.,
Breast cancer tumor growth is efficiently inhibited by dendritic cell transfusion in murine model. Biomedical Research and Therapy.
2014;
1
(03)
:
85-92
.
View Article Google Scholar -
Wu
J.,
Wu
H.,
An
J.,
Ballantyne
C.M.,
Cyster
J.G.,
Critical role of integrin CD11c in splenic dendritic cell capture of missing-self CD47 cells to induce adaptive immunity. Proceedings of the National Academy of Sciences of the United States of America.
2018;
115
(26)
:
6786-91
.
View Article PubMed Google Scholar -
Aerts-Toegaert
C.,
Heirman
C.,
Tuyaerts
S.,
Corthals
J.,
Aerts
J.L.,
Bonehill
A.,
CD83 expression on dendritic cells and T cells: correlation with effective immune responses. European Journal of Immunology.
2007;
37
(3)
:
686-95
.
View Article PubMed Google Scholar -
Iorns
E.,
Drews-Elger
K.,
Ward
T.M.,
Dean
S.,
Clarke
J.,
Berry
D.,
A new mouse model for the study of human breast cancer metastasis. PLoS One.
2012;
7
(10)
:
e47995
.
View Article PubMed Google Scholar -
Dhodapkar
M.V.,
Dhodapkar
K.M.,
Palucka
A.K.,
Interactions of tumor cells with dendritic cells: balancing immunity and tolerance. Cell Death and Differentiation.
2008;
15
(1)
:
39-50
.
View Article PubMed Google Scholar -
Melief
C.J.,
Cancer immunotherapy by dendritic cells. Immunity.
2008;
29
(3)
:
372-83
.
View Article PubMed Google Scholar -
Gerner
M.Y.,
Casey
K.A.,
Mescher
M.F.,
Defective MHC class II presentation by dendritic cells limits CD4 T cell help for antitumor CD8 T cell responses. Journal of Immunology (Baltimore, Md.: 1950).
2008;
181
(1)
:
155-64
.
View Article PubMed Google Scholar -
McDonnell
A.M.,
Prosser
A.C.,
van Bruggen
I.,
Robinson
B.W.,
Currie
A.J.,
CD8alpha+ DC are not the sole subset cross-presenting cell-associated tumor antigens from a solid tumor. European Journal of Immunology.
2010;
40
(6)
:
1617-27
.
View Article PubMed Google Scholar -
Shortman
K.,
Heath
W.R.,
The CD8+ dendritic cell subset. Immunological Reviews.
2010;
234
(1)
:
18-31
.
View Article PubMed Google Scholar -
Itano
A.A.,
Jenkins
M.K.,
Antigen presentation to naive CD4 T cells in the lymph node. Nature Immunology.
2003;
4
(8)
:
733-9
.
View Article PubMed Google Scholar -
Hildner
K.,
Edelson
B.T.,
Purtha
W.E.,
Diamond
M.,
Matsushita
H.,
Kohyama
M.,
Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science.
2008;
322
(5904)
:
1097-100
.
View Article PubMed Google Scholar -
Wright
S.E.,
Immunotherapy of breast cancer. Expert Opinion on Biological Therapy.
2012;
12
(4)
:
479-90
.
View Article PubMed Google Scholar -
Ueno
N.T.,
Rizzo
J.D.,
Demirer
T.,
Cheng
Y.C.,
Hegenbart
U.,
Zhang
M.J.,
Allogeneic hematopoietic cell transplantation for metastatic breast cancer. Bone Marrow Transplantation.
2008;
41
(6)
:
537-45
.
View Article PubMed Google Scholar -
Sparano
J.A.,
Fisher
R.I.,
Weiss
G.R.,
Margolin
K.,
Aronson
F.R.,
Hawkins
M.J.,
Phase II trials of high-dose interleukin-2 and lymphokine-activated killer cells in advanced breast carcinoma and carcinoma of the lung, ovary, and pancreas and other tumors. Journal of Immunotherapy with Emphasis on Tumor Immunology.
1994;
16
(3)
:
216-23
.
View Article PubMed Google Scholar -
Bernhard
H.,
Neudorfer
J.,
Gebhard
K.,
Conrad
H.,
Hermann
C.,
Nährig
J.,
Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunology, Immunotherapy.
2008;
57
(2)
:
271-80
.
View Article PubMed Google Scholar -
Gao
G.,
Wang
Z.,
Qu
X.,
Zhang
Z.,
Prognostic value of tumor-infiltrating lymphocytes in patients with triple-negative breast cancer: a systematic review and meta-analysis. BMC Cancer.
2020;
20
(1)
:
179
.
View Article PubMed Google Scholar -
Denkert
C.,
von Minckwitz
G.,
Darb-Esfahani
S.,
Lederer
B.,
Heppner
B.I.,
Weber
K.E.,
Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. The Lancet. Oncology.
2018;
19
(1)
:
40-50
.
View Article PubMed Google Scholar -
Ali
H.R.,
Provenzano
E.,
Dawson
S.J.,
Blows
F.M.,
Liu
B.,
Shah
M.,
Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Annals of Oncology : Official Journal of the European Society for Medical Oncology.
2014;
25
(8)
:
1536-43
.
View Article PubMed Google Scholar -
West
N.R.,
Milne
K.,
Truong
P.T.,
Macpherson
N.,
Nelson
B.H.,
Watson
P.H.,
Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Research.
2011;
13
(6)
:
126
.
View Article PubMed Google Scholar -
Wylie
B.,
Macri
C.,
Mintern
J.D.,
Waithman
J.,
Dendritic cells and cancer: from biology to therapeutic intervention. Cancers (Basel).
2019;
11
(4)
:
521
.
View Article PubMed Google Scholar -
Neidhardt-Berard
E.M.,
Berard
F.,
Banchereau
J.,
Palucka
A.K.,
Dendritic cells loaded with killed breast cancer cells induce differentiation of tumor-specific cytotoxic T lymphocytes. Breast Cancer Research.
2004;
6
(4)
:
322-8
.
View Article PubMed Google Scholar -
Wang
B.,
Zaidi
N.,
He
L.Z.,
Zhang
L.,
Kuroiwa
J.M.,
Keler
T.,
Targeting of the non-mutated tumor antigen HER2/neu to mature dendritic cells induces an integrated immune response that protects against breast cancer in mice. Breast Cancer Research.
2012;
14
(2)
:
39
.
View Article PubMed Google Scholar -
Sakai
S.,
Kauffman
K.D.,
Sallin
M.A.,
Sharpe
A.H.,
Young
H.A.,
Ganusov
V.V.,
CD4 T cell-derived IFN-γ plays a minimal role in control of pulmonary Mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathogens.
2016;
12
(5)
:
e1005667
.
View Article PubMed Google Scholar -
Banchereau
J.,
Schuler-Thurner
B.,
Palucka
A.K.,
Schuler
G.,
Dendritic cells as vectors for therapy. Cell.
2001;
106
(3)
:
271-4
.
View Article PubMed Google Scholar -
Fuentes-Antrás
J.,
Guevara-Hoyer
K.,
Baliu-Piqué
M.,
García-Sáenz
J.Á.,
Pérez-Segura
P.,
Pandiella
A.,
Adoptive cell therapy in breast cancer: A current perspective of next-generation medicine. Frontiers in Oncology.
2020;
10
:
605633
.
View Article PubMed Google Scholar -
Soysal
S.D.,
Tzankov
A.,
Muenst
S.E.,
Role of the tumor microenvironment in breast cancer. Pathobiology.
2015;
82
(3-4)
:
142-52
.
View Article PubMed Google Scholar -
Luckheeram
R.V.,
Zhou
R.,
Verma
A.D.,
Xia
B.,
CD4⁺T cells: differentiation and functions. Clinical & Developmental Immunology.
2012;
2012
:
925135
.
View Article PubMed Google Scholar -
Benevides
L.,
da Fonseca
D.M.,
Donate
P.B.,
Tiezzi
D.G.,
De Carvalho
D.D.,
de Andrade
J.M.,
IL17 promotes mammary tumor progression by changing the behaviour of tumor cells and eliciting tumorigenic neutrophils recruitment. Cancer Research.
2015;
75
(18)
:
3788-99
.
View Article PubMed Google Scholar -
Gupta
S.,
Joshi
K.,
Wig
J.D.,
Arora
S.K.,
Intratumoral FOXP3 expression in infiltrating breast carcinoma: its association with clinicopathologic parameters and angiogenesis. Acta Oncologica (Stockholm, Sweden).
2007;
46
(6)
:
792-7
.
View Article PubMed Google Scholar -
Pan
K.,
Zhao
J.J.,
Wang
H.,
Li
J.J.,
Liang
X.T.,
Sun
J.C.,
Comparative analysis of cytotoxic T lymphocyte response induced by dendritic cells loaded with hepatocellular carcinoma -derived RNA or cell lysate. International Journal of Biological Sciences.
2010;
6
(7)
:
639-48
.
View Article PubMed Google Scholar -
Sumransub
N.,
Jirapongwattana
N.,
Jamjuntra
P.,
Thongchot
S.,
Chieochansin
T.,
Yenchitsomanus
P.T.,
Breast cancer stem cell RNA-pulsed dendritic cells enhance tumor cell killing by effector T cells. Oncology Letters.
2020;
19
(3)
:
2422-30
.
View Article PubMed Google Scholar -
Garg
N.K.,
Dwivedi
P.,
Prabha
P.,
Tyagi
R.K.,
RNA pulsed dendritic cells: an approach for cancer immunotherapy. Vaccine.
2013;
31
(8)
:
1141-56
.
View Article PubMed Google Scholar
Comments
Article Details
Volume & Issue : Vol 9 No 5 (2022)
Page No.: 5051-5064
Published on: 2022-05-31
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
- HTML viewed - 5121 times
- PDF downloaded - 1557 times
- XML downloaded - 0 times
