

Copyrights: Ha Thi Chu, Doanh Huu Le, Thieu Van Le, Binh Bui Nguyen, Janice Schwartz, Quang Van Vu, 2021. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Background: Pachyonychia congenita (PC) comprises a group of rare autosomal dominant genetic disorders. It is characterized by hypertrophic nail dystrophy, focal palmoplantar keratoderma, follicular keratosis, and oral leukokeratosis. It is associated with mutations in five differentiationspecific keratin genes: KRT6A, KRT6B, KRT6C, KRT16, or KRT17. The case is being reported for its rarity. To the best of our knowledge, this is the first report of PC, from Vietnam, confirmed by genetic analysis.
Case presentation: A four-year-old Vietnamese girl presented with a thickened nail and oral leukokeratosis soon after birth. She was diagnosed with onychomycosis and chronic oral candidiasis and was treated with systemic anti-fungals in children's hospitals and dermatology departments multiple times; however, no treatments were effective. In collaboration with the International Pachyonychia Congenita Research Registry (IPCRR), the clinical features were consistent with a diagnosis of PC type PC-K6a. The genetic testing, performed through the IPCRR, shows a K6a N171K mutation.
Conclusions: The IPCRR helps screen PC's clinical features and confirm a diagnosis at the molecular level, which is beneficial and crucial for validating the condition's clinical impression.
Introduction
Pachyonychia congenita (PC) is a rare autosomal dominant disorder of keratinization. It is characterized by hypertrophic nail dystrophy, follicular hyperkeratosis, painful plantar and palmar keratoderma, oral leukokeratosis, epidermal cysts, natal teeth, and hoarseness1. Genetic testing of PC is critical for diagnostic confirmation and genetic counseling1, 2. The International Pachyonychia Congenita Research Registry (IPCRR, WCG IRB #20040468), sponsored by Pachyonychia Congenita Project, was formulated in 2004 and since then has collected data on over 2,000 patients suspected of having PC and provides free genetic testing for those in the IPCRR. Research based on the IPCRR data has shown significant overlap between previous types of PC. Thus, a new classification was proposed in 2011 that grouped PC into five types: PC-K6a, PC-K6b, PC-K6c, PC-K16, and PC-K17, which represents specific affected genes3. This is the classification now used to describe PC. Underlying keratin gene mutations have been described in the five keratin genes, KRT6A (OMIM 148041), KRT6B (OMIM 148042), KRT6C (OMIM 612315), KRT16 (OMIM 148067), and KRT17 (OMIM 148069), which alter keratins 6a, 6b, 6c, 16, and 17, respectively4. These genes are expressed in the nail bed, palmoplantar epidermis, and mucosa. Keratins play a key role in epidermal cell integrity and mechanical strength, and mutations in any of these five keratin genes cause epidermolysis and compensatory hyperkeratosis at these sites5.
Here, we report a case of this rare disease. To the best of our knowledge, this is the first PC case from Vietnam confirmed by genetic analysis.

Case presentation
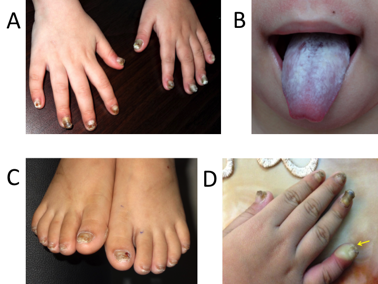
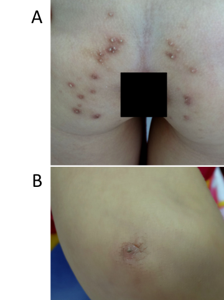
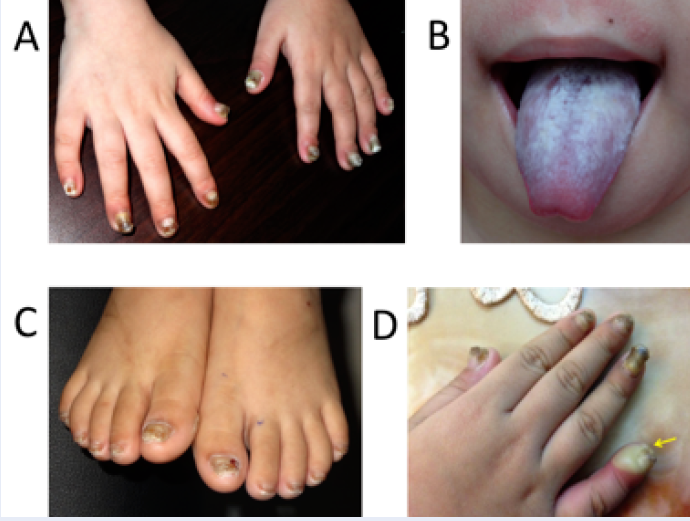
A four-year-old female patient was presented at our hospital (March 2016) because of leukokeratosis and thickened nails with a progressive course since early infancy. She is the first child of nonconsanguineous parentage with no history of genetic disease. She underwent an uncomplicated birth and all normal developmental milestones for her age. Her mother noticed nail changes at 15 days and the leukokeratotic plaques at 30 days. The child was diagnosed with onychomycosis, candida infection and was treated with local, systemic anti-fungals at pediatric and dermatology hospitals; however, all treatments were ineffective. A physical examination identified a distinct hypertrophic nail dystrophy of the feet and hands, with distal hyperkeratosis and curvature of nails especially on the fingers, which were wedge-shaped with yellowish discoloration (Figure 1 A, C, D). Sometimes, the fingernails would become infected and fall off (Figure 1 D). Oral leukokeratosis covered the entire dorsal aspect of her tongue and fluctuated with time (Figure 1 B). She had focal plantar keratoderma, but at the time, it did not affect her walking or limit her mobility. The hoarseness of her voice was not reported. Hyperkeratotic papules appeared over her buttocks (Figure 2 A) and elbows (Figure 2 B). Hyperkeratotic papules were exacerbated by warm and dry weather. Topical keratolytics and emollients were prescribed but showed limited benefit. Routine laboratory investigations including complete hemogram, hepatic profile, and renal profile were within normal limits. Histologically, we identified the marked proliferation of keratin with an edematous change; no Aspergillus infection was observed in the nail specimens (Figure 3 A-B). This was also confirmed by immunohistochemical staining using an anti-Aspergillus antibody. No evidence of any malignancy was found during the comprehensive examination.
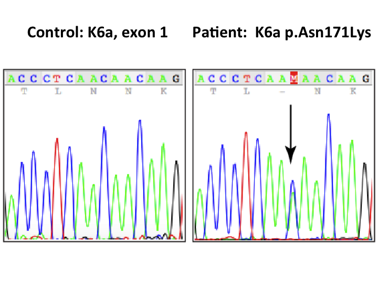
Based on the above findings, the patient was clinically diagnosed with PC. After being referred to the PC Project (www.pachyonychia.org), a saliva kit for genetic testing was freely provided by the PC Project. PC Project offers genetic testing to PC patients who enroll in the IPCRR (https://www.pachyonychia.org/patient-registry/). Following the acquisition of informed consent according to the Declaration of Helsinki principles, genetic testing was performed to confirm the clinical diagnosis. The results revealed the KRT6A gene to have the recurring mutation at N171K. Because of the mutation, one of the amino acid building blocks Asparagine (Asn or N) changed to Lysin (Lys or K); therefore, the correct protein is not produced.
After being diagnosed with PC, the patient was no longer mistreated. We suggested appropriate care and therapeutic solutions to improve her quality of life.
Discussion & Conclusions
Pachyonychia Congenita is a rare autosomal dominant keratin disorder caused by mutations in any of the five keratin genes: KRT6A, KRT6B, KRT6C, KRT16, or KRT176. The major phenotypical features of PC reported in more than 90% of patients with this condition are nail dystrophy, plantar keratoderma, and plantar pain. Other common clinical findings of PC include oral leukokeratosis, follicular hyperkeratosis, and the formation of cysts1, 7. Additionally, natal teeth, a phenomenon of erupted teeth present at birth, have also been reported in a few patients with PC-K17. Based on clinical characteristics, PC was classified into two primary subtypes: the Jadassohn-Lewandowski PC type 1 (mutations in KRT 6A and KRT16) and Jackson-Lawler PC type 2 (mutations in KRT6B and KRT 17). Because of a significant phenotype overlap between cases previously classified as PC types 1 and 2, a new classification divided PC into four subtypes based on the specific keratin defect1, 6, 7 and later into five subtypes. PC subtypes are now classified as PC-K6a, PC-K6b, PC-K6c, PC-K16, and PC-K17 for mutations in KRT6A, KRT6B, KRT6C, KRT16, and KRT17 genes, respectively. Due to the overlapping clinical features between the different subtypes of PC, genetic testing is required to confirm the diagnosis2. The patient we herein report showed typical manifestations including thickened nails, oral leukocytosis, hyperkeratotic papules, and focal plantar keratoderma soon after birth. However, the diagnosis was missed and delayed until the age of four. Her nail dystrophy and oral leukokeratosis were diagnosed as onychomycosis and chronic candidiasis, respectively. This misdiagnosis can be explained by the following reasons: there is insufficient awareness of this rare disease, and Vietnam is a tropical country, where infectious diseases including fungal diseases are common8, 9. In our hospital, the patient also was first suspected of having onychomycosis. The nail culture also showed Aspergillus versicolor; however, was this fungus pathogenesis or merely superinfection? Histologically, no finding of Aspergillus versicolor in the nail specimens examined was seen but there was marked proliferation of keratin. No evidence of malignancy was observed either. Therefore, a diagnosis of PC was made. Genetic testing was performed that revealed the K6a N171K mutation, thereby confirming the clinical diagnosis. This mutation has been previously reported in 18 individuals in the world10. Our patient represents one of more than 45% of PC cases that appear spontaneously with no family history (de novo mutation).
According to data of PC project, there were 977 PC individuals in 517 families worldwide confirmed by genetic analysis; 115 mutations across the five keratin genes were identified. The percentage of KRT6A, KRT6B, KRT6C, KRT16, and KRT17 gene mutations is 39, 9, 3, 33, and 16, respectively. PC-K6a affects the largest group of PC patients characterized by early-onset and extensive nail dystrophy; severe, painful plantar keratoderma; follicular hyperkeratosis; and oral leukokeratosis10, 11. While nearly 100% of PC-K6a patients over six years old have painful plantar keratoderma that negatively impacts quality of life, our patient seldom has pain. This may be explained by her young age. After the confirmed diagnosis, the patient is no longer mistreated. Although there is no specific treatment for PC, the supportive therapies available on the PC Project website have proven effective to improve the daily activities of our patient. We continue to support the patient to improve her quality of life. The experience from this first PC case has helped us provide an early diagnose to many other PC patients scattered throughout Vietnam. In summary, we report the first case of PC-K6a confirmed by genetic analysis from Vietnam under the support of the PC Project. This study stressed the importance of genetic testing to confirm the diagnosis and genetic counseling.
Abbreviations
PC: Pachyonychia congenita
IPCRR: International Pachyonychia Congenita Research Registry
Acknowledgments
The authors would like to thank the patient and her family in this study for their cooperation. Thanks also to PC project including Holly Evans for her help with data collection to the International Pachyonychia Congenita Consortium Genetics Team for useful suppor.
Author’s contributions
HTC, QVV, DHL, TVL and BBN participated in the study design, protocol development and performance, data analysis, interpretation of data, and writing of the manuscript and carried out the clinical data collection and data analysis. QVV and JS reviewed and revised the manuscript, making important intellectual contributions. All authors read and approved the final manuscript.
Funding
Genetic testing work was supported by Pachyonychia Congenita Project.
Availability of data and materials
Data and materials used and/or analysed during the current study are available from the corresponding author on reasionable request.
Ethics approval and consent to participate
This study was conducted in accordance with the amended Declaration of Helsinki. Written informed consent was obtained from the patient and his parents for publication of these data and for the accompanying images.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
-
Eliason
M.J.,
Leachman
S.A.,
Feng
B.J.,
Schwartz
M.E.,
Hansen
C.D.,
A review of the clinical phenotype of 254 patients with genetically confirmed pachyonychia congenita. J Am Acad Dermatol.
2012;
67
(4)
:
680-6
.
View Article PubMed Google Scholar -
Rusu
C.,
Murgu
A.,
Chiriac
A. E.,
Wilson
N. J.,
Smith
F. J. D.,
First Report of Pachyonychia Congenita Type PC-K6a in the Romanian Population. Maedica (Buchar).
2017;
12
(2)
:
123-6
.
PubMed Google Scholar -
McLean
W.H.,
Hansen
C.D.,
Eliason
M.J.,
Smith
F.J.,
The phenotypic and molecular genetic features of pachyonychia congenita. J Invest Dermatol.
2011;
131
(5)
:
1015-7
.
View Article PubMed Google Scholar -
Pachyonychia Congenita - GeneReviews® - NCBI Bookshelf [Internet]. [cited 2018 Aug 22]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1280/.
.
-
Shah
S.,
Boen
M.,
Kenner-Bell
B.,
Schwartz
M.,
Rademaker
A.,
Paller
A.S.,
Pachyonychia congenita in pediatric patients: natural history, features, and impact. JAMA Dermatol.
2014;
150
(2)
:
146-53
.
View Article PubMed Google Scholar -
Wilson
N.J.,
Leachman
S.A.,
Hansen
C.D.,
McMullan
A.C.,
Milstone
L.M.,
Schwartz
M.E.,
A large mutational study in pachyonychia congenita. J Invest Dermatol.
2011;
131
(5)
:
1018-24
.
View Article PubMed Google Scholar -
Ghazawi
F.M.,
Hassani-Ardakani
K.,
Henriques
L.,
Jafarian
F.,
Identification of a novel substitution mutation (R103C) in the rod domain of the keratin 17 gene associated with pachyonychia congenita type 2. Int J Dermatol.
2018;
58
(2)
:
233-236
.
View Article PubMed Google Scholar -
Vu
Q.V.,
Acne fulminans in a Vietnamese boy successfully treated with prednisolone and ibuprofen: A case report. Biomed Res Ther.
2018;
5
(9)
:
2708-2711
.
View Article Google Scholar -
Tran
T.T.,
Vu
Q.V.,
Wada
T.,
Yachie
A.,
Le Thi Minh
H.,
Nguyen
S.N.,
Novel HAX1 Gene Mutation in a Vietnamese Boy with Severe Congenital Neutropenia. Case Rep Pediatr.
2018;
2018
:
2798621
.
View Article PubMed Google Scholar -
PC Data [Internet]. [cited 2021 Feb 26]. Available from: https://www.pachyonychia.org/pc-data/.
.
-
Forrest
C.E.,
Casey
G.,
Mordaunt
D.A.,
Thompson
E.M.,
Gordon
L.,
Pachyonychia Congenita: A Spectrum of KRT6a Mutations in Australian Patients. Pediatr Dermatol.
2016;
33
(3)
:
337-42
.
View Article PubMed Google Scholar
Comments
Downloads
Article Details
Volume & Issue : Vol 8 No 6 (2021)
Page No.: 4434-4438
Published on: 2021-06-30
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 6220 times
- Download downloaded - 1892 times
- View Article downloaded - 0 times
