

Copyrights: Rozhgar A. Khailany, Naser Gilani, Mehmet Ozaslan, Muhamad Safdar, Ihsan Al-Shamari, Belan O. Kanabe, Khandakar A. S. M. Saadat, Javad Homayounvash, Amir Monfaredan, Mustafa S. Al-Attar, Ahmet Arslan, 2020. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Leigh Syndrome (LS) is an uncommon progressive neurodegenerative mitochondrial disorder. The condition is characterized by progressive mental and developmental disabilities (psychomotor regression) and commonly brings about death within a few years of diagnosis, more often due to respiratory failure. In a small number of patients the disorder does not manifest until adulthood. The principal indications of Leigh syndrome found in early stages typically are diarrhea, vomiting, and difficulty swallowing (dysphagia), which disturbs eating. These problems usually result in powerlessness to develop and put on weight under the normal rate (failure to thrive). Serious movement and muscle problems are basic in Leigh syndrome. In this case report, we introduce the molecular and clinical features of a 19-year-old female as proband, and also, we study other members of the family consequently. The m.9176T>G heteroplasmic mutation in the MT-ATP6 gene was detected by high-resolution melt (HRM) and DNA sequencing techniques. Similarly, the m.9176T>G was heteroplasmic in the mother. In conclusion, this report in compliance with previous studies underlines the necessity of further research on prenatal distinguishing proof of the responsible mutations and avoidance of the disease in families with known cases.
INTRODUCTION
Leigh Syndrome (LS), also known as subacute necrotizing encephalomyopathy (SNEM) is an uncommon inherited, mitochondrial DNA-associated, and severe neurodegenerative disease which is marked by bilaterally symmetrical necrotic lesions in the brainstem and basal ganglia1, 2. Typically, disease onset occurs between 3 and 12 months of age and progresses to death within two years2. Prognosis is poor, but late-onset and slower progression have also been observed2, 3. LS was first described by Denis Leigh, a British neuropathologist in 1951, in a 7-month-old infant with a rapidly progressive form of the disease that resulted in death over six weeks4. LS is caused by abnormalities in mitochondrial energy generation5. Clinically, patients manifest a heterogeneous set of symptoms, including psychomotor regression (loss of mental and movement abilities), muscular hypotonia, ataxia, dystonia, respiratory insufficiency, and brainstem signs (strabismus, nystagmus and swallowing difficulties)1, 2, 3.
Genetically, LS is a highly heterogeneous mitochondrial disease5. New genetic defects are being increasingly recognized in light of novel techniques of whole-genome and next-generation sequencing5. Investigation of causative genetic defects could be a cumbersome undertaking, as researchers are confronted with two distinguished genomes- mitochondrial, and nuclear5, 6. LS may be caused by defects in more than 35 different genes of either nuclear or mitochondrial origin5. Nevertheless, the exact genetic defects, in many cases, remain unknown5.
Mitochondrially-encoded ATP synthase 6 (MT-ATP6) gene, also known as ATPase-6, is a key enzyme of mitochondrial energy conversion6. It is situated in mitochondrial DNA (mtDNA)- nucleotide bases 8527 to 9207- and belongs to a family of genes termed mitochondrial respiratory chain complex7. Approximately 10–20% of individuals with LS have a mutation in mtDNA of the MT-ATP6 gene, with the most common genetic alteration being 8993T>G8. The syndromes of LS and muscle weakness, ataxia and retinitis pigmentosa (NARP), named ‘striatal necrosis syndromes’, are associated with mutations at nucleotide position 9176 of the MT-ATP6 gene8. The MT-ATP6 defects that lead to LS impair the stability or function of the ATP synthase complex, suppressing ATP production and damaging oxidative phosphorylation8.
Here, we present the molecular and clinical features of a 19-year-old female as proband affected by LS associated with the mtDNA defect, and we also consequently evaluate other members of the family.


CASE PRESENTATION
The proband is a 19 years old female patient born to consanguineous Arab parents after a regular pregnancy and delivery, referred to genetic laboratory by a Neurologist. Her parents are first cousins (Figure 1); they are an Arab family from the Anbar Province of Iraq living in Erbil as war refugees. Phenotypically, the parents are normal, with no signs and symptoms of LS. Upon evaluation, the proband had retinitis pigmentosa, progressive dementia, and muscle weakness. Her problems started when she was in her early twenties, with movement disabilities and muscle weakness, followed by hearing and vision problems; after that, she could not speak normally. There was also a history of seizures.


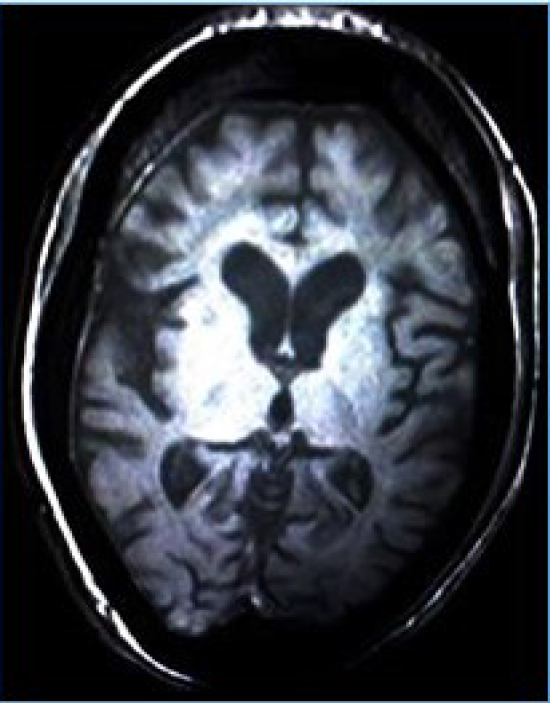
On ultrasound of ophthalmic cavities, there were a few thin bands in the left vitreous cavity, and the posterior wall of both globes was thick with the irregular inner surface. The electroencephalography (EEG) examination showed short runs and frequent bursts of bilateral synchronous spikes, polyspikes, sharp waves, slow-wave complexes, and phase reversal. Magnetic resonance imaging (MRI) showed slightly dilated cerebrospinal fluid (CSF) spaces, suggestive of diffuse brain atrophic changes (Figure 2). Unfortunately, before we finished her evaluations, she died due to respiratory failure.
On molecular investigations, we studied four different known mutations as follows: 3243A>G in the MT-TL1 gene, 8344A>G in the MT-TK gene, as well as m.9191T>C and m.9176T>G in the MT-ATP6 gene. The mutations were assessed by monitoring high-resolution melt (HRM) and nucleotide sequencing techniques. The case had m.9176T>G mutation in the MT-ATP6 gene (Figure 3, Figure 4), resulting in an amino acid substitution of Leu to Arg of ATPase-6. However, there were no mutations in the other three locations.
The family has a history of early childhood death of their four offspring (three girls and one boy); three of them passed away in their first year of life and one passed away in her 4th year. All of them died due to respiratory failure. The parents had five live offspring, including four girls (the proband passed away in the course of our investigations), and one boy (three of them have abnormal phenotypes). After the detection of a mutation in MT-ATP6 in the proband, we decided to study all family members by molecular analyses. The clinical characteristics and molecular investigations of the living children and their mother are shown in Table 1. Notably, the mtDNA investigations of the mother at that position demonstrated that she was heteroplasmic for the mutation.
| Family Member | Clinical Findings | Molecular Investigation |
|---|---|---|
| Mother | Asymptomatic | MT-ATP6 (m.9176T>G) detected |
| 20 months old girl | General muscular weakness, hearing loss, hypotonia, dysphasia, limited movements | MT-ATP6 (m.9176T>G) detected |
| 5 years old girl | Vision problem in darkness from her 3 years of age, there is no other sign and symptom | MT-ATP6 (m.9176T>G) detected |
| 12 years old blind girl | Her vision problem started when she was 9 years old and finally lead to complete blindness in 11 years of age, normal mental function, normal neuromuscular examination | MT-ATP6 (m.9176T>G) detected |
| 18 years old boy | No history of any kind of mental and movement disability, normal neuromuscular exam, plays football sometimes | MT-ATP6 (m.9176T>G) not detected |
DISCUSSION
Leigh syndrome is an uncommon inherited, neurometabolic, subacute necrotizing encephalopathy that affects the central nervous system, and occurs in 1 of 40,000 newborns worldwide and 1 of 2,000 newborns in certain populations of Saguenay Lac-Saint-Jean region of Quebec, Canada 3. Leigh syndrome is a serious, multisystem and progressive metabolic neurodegenerative disorder, with distinct neuroradiological and neuropathological alterations as well as prominent clinical and genetic heterogeneity5.
With current genetic diagnostic technology, Leigh syndrome has been diagnosed to be the result of mutations in more than 35 distinct genes of either nuclear or mitochondrial origin, including in each of the five respiratory chain complexes5.
In the study herein, we detected a point substitution mutation of the MT-ATP6 gene (m.9176T>G) in a patient from Iraq. Similarly, Motohiro et al. (2012) reported the same mutation of ATP6 in Leigh syndrome9.
The clinicopathological features of the mutated case include retinitis pigmentosa, progressive dementia, and muscle weakness. MT-ATP6 is one of the mitochondrial genes that is involved in ATP production through the oxidative phosphorylation process and mutation (m.9176T>G) of this gene results in a defect at complex V, and subsequently harms the capacity of the proton channel, resulting in loss of ATP-synthetic activity10. Alteration of this gene gives rises to the most well-known maternally-inherited Leigh syndrome (MILS), as the most widespread mtDNA mutation in Leigh syndrome5.
The ATP synthase enzyme is a multi-subunit complex with a molecular mass of around 550,000 Da5. It has both a hydrophilic ATPase and a hydrophobic domain5. ATP synthase deformities might be of either nuclear or mitochondrial genetic origin since the biogenesis of the mitochondrial ATP synthase has 14 subunits that are nuclear-encoded, in addition to 2 mtDNA-encoded proteins5.
Regarding the mutation, Carrozzo et al. has found inactive ATP synthetase in E. coli9. From this evidence, the mutation is believed to be a neurotic and vital part of the amino acid leucine at that position in ATPase11. Along with a mtDNA mutation at nucleotide 8993, having a T-to-G or T-to-C mutation provides solid proof to the significance of ATP synthetase brokenness in maternally-inherited Leigh syndrome9, 10.
CONCLUSION
Leigh syndrome is a genetically heterogeneous disorder characterized by a vast spectrum of phenotypes and a variable disease course. This report corroborates previous studies since our findings in this case report comply with other case reports of this rare syndrome. Particularly, in this crowded family with nine kids, we observed considerable clinical variability among the siblings since 5 of them had passed away, while the 3 living sibling show a different clinical picture. Finally, there is a clinically normal 18-year-old male offspring. Our case report may provide a rationale for examinations utilizing cells or tissues from these patients in order to identify novel genes and novel mechanisms involved in the maintenance and assembly of the mitochondrial complex in this syndrome.
Abbreviations
MT-ATP6: Mitochondrially encoded ATP synthase membrane subunit 6
LS: Leigh Syndrome
HRM: High-resolution melting
SNEM: Subacute necrotizing encephalomyopathy
mtDNA: Mitochondrial DNA
NARP: Neuropathy, ataxia, and retinitis pigmentosa
ATP: Adenosine triphosphate
EEG: Electroencephalography
MRI: Magnetic resonance imaging
CSF: Cerebrospinal fluid
MT-TL1: Mitochondrially encoded tRNA leucine 1
MILS: Maternally inherited Leigh syndrome
Acknowledgments
Not applicable.
Author’s contributions
Rozhgar A. Khailany and Naser Gilani: Conceived and designed the experiments, Rozhgar A. Khailany, Naser Gilani, Mehmet Ozaslan, Muhamad Safdar: preparation and article reviewing, Rozhgar A. Khailany, Naser Gilani and Amir Monfaredan: performed the experiments, Ihsan Al-Shamari: Clinical presentation, Mehmet Ozaslan, Muhamad Safdar, Belan O. Kanabe, Khandakar A. S. M. Saadat, Javad Homayounvash, Mustafa S. Al-Attar and Ahmet ARSLAN: Interpretation and drafting article and revising it critically. All authors read and approved the manuscript.
Funding
Not applicable.
Availability of data and materials
Data and materials used and/or analysed during the current study are available from the corresponding author on reasionable request.
Ethics approval and consent to participate
This study was conducted in accordance with the amended Declaration of Helsinki. The institutional review board approved the study (approval number: 3/1/1011- 2016), and all participants provided written informed consent.
Consent for publication
The author hereby consents that the publisher publishes the work.
Competing interests
The authors declare that they have no competing interests.
References
-
Shrikhande
D.Y.,
Kotalaki
P.,
Syed
M.M.A.,
Ahya
K.,
Singh
G.,
A rare mitochondrial disorder: Leigh syndrome - a case report. Italian Journal of Pediatrics.
2010;
36
:
62
.
View Article PubMed Google Scholar -
Ronchi
D.,
Cosi
A.,
Tonduti
D.,
Orcesi
S.,
Bordoni
A.,
Fortunato
F.,
Rizzuti
M.,
Sciacco
M.,
Collotta
M.,
Cagdas
S.,
Capovilla
G.,
Moggio
M.,
Berardinelli
A.,
Veggiotti
P.,
Comi
G.P.,
Clinical and molecular features of an infant patient affected by Leigh Disease associated to m.14459G > A mitochondrial DNA mutation: a case report. BMC Neurology.
2011 ;
11
:
85
.
View Article PubMed Google Scholar -
Dinesh
P.,
Raj
M.M.,
Gita
S.,
Leigh Syndrome: An Unusual Rare Case Report. Int J Sci Stud.
2014;
2
(2)
:
93-96
.
-
Horva'th
R.,
Abicht
A.,
Holinski-Feder
E.,
Laner
A.,
Gempel
K.,
Prokisch
H.,
Lochmu¨ller
H.,
Klopstock
T.,
Jaksch
M.,
Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA). J Neurol Neurosurg Psychiatry.
2006;
77
:
74-76
.
View Article PubMed Google Scholar -
Akagi
M.,
Inui
K.,
Tsukamoto
H.,
Sakai
N.,
Muramatsu
T.,
Yamada
M.,
Matsuzaki
K.,
Goto
Y.,
Nonaka
I.,
Okada
S.,
A point mutation of mitochondrial ATPase 6 gene in Leigh syndrome. Neuromuscul Disord.
2002;
12
:
53-55
.
View Article Google Scholar -
Houštěk
J.,
Pícková
A.,
Vojtíšková
A.,
Mráček
T.,
Pecina
P.,
Ješina
P.,
Mitochondrial diseases and genetic defects of ATP synthase. Biochimica et Biophysica Acta.
2006;
1757
:
1400-1405
.
View Article PubMed Google Scholar -
https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=4508.
.
-
pez-Gallardo1
E.L.,
Emperador1
S.,
Solano1
A.,
Llobet1
L.,
Martı'n-Navarro1
A.,
pez-Pe'rez1
M.J.L,
Briones
P.,
Pineda
M.,
Artuch
R.,
Barraquer
E.,
Jerico'
I.,
Ruiz-Pesini1
E.,
Montoya
J.,
Expanding the clinical phenotypes of MT-ATP6 mutations. Human Molecular Genetics.
2014;
23
(23)
:
6191-6200
.
View Article PubMed Google Scholar -
Akagia
M.,
Inuia
K.,
Tsukamotoa
H.,
Sakaia
N.,
Muramatsua
T.,
Yamadaa
M.,
Matsuzakib
K.,
Gotoc
Y.,
Nonakad
I.,
Okadaa
S.,
A point mutation of mitochondrial ATPase 6 gene in Leigh syndrome. Neuromuscular Disorders.
2002;
12
:
53-55
.
View Article Google Scholar -
Tatuch
Y.,
Robinson
B.H.,
The mitochondrial DNA mutation at 8993 associated with NARP slows the rate of ATP synthesis in isolated lymphoblast mitochondria. Biochem Biophys Res Commun.
1993;
192
(1)
:
124-128
.
View Article PubMed Google Scholar -
Dahl
H.H.M.,
Getting to the nucleus of mitochondrial disorders: identification of respiratory chain-enzyme genes causing Leigh syndrome. Am J Hum Genet.
1998;
63
:
1594-1597
.
View Article PubMed Google Scholar
Comments
Downloads
Article Details
Volume & Issue : Vol 7 No 5 (2020)
Page No.: 3739-3743
Published on: 2020-05-25
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
- HTML viewed - 6750 times
- Download PDF downloaded - 1763 times
- View Article downloaded - 0 times
