

Copyrights: Abolfazl Nasiri, Masoud Sadeghi, Asad Vaisi-Raygani, Sara Kiani, Zahra Aghelan, Reza Khodarahmi, 2020. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
The pyruvate dehydrogenase complex (PDC) is a multi-enzyme complex of the mitochondria that provides a link between glycolysis and the Krebs cycle. PDC plays an essential role in producing acetyl-CoA from glucose and the regulation of fuel consumption. In general, PDC enzyme is regulated in two different ways, end-product inhibition and posttranslational modifications (more extensive phosphorylation and dephosphorylation subunit E1). Posttranslational modifications of this enzyme are regulated by various factors. Sirtuins are the class III of histone deacylatases that catalyze protein posttranslational modifications, including deacetylation, adenosine diphosphate ribosylation, and deacylation. Sirt3, Sirt4, and Sirt5 are mitochondrial sirtuins that control the posttranslational modifications of mitochondrial protein. Considering the comprehensive role of sirtuins in post-translational modifications and regulation of metabolic processes, the aim of this review is to explore the role of mitochondrial sirtuins in the regulation of the PDC. PDC deficiency is a common metabolic disorder that causes pyruvate to be converted to lactate and alanine rather than to acetyl-CoA. because this enzyme is in the gateway of complete oxidation, glucose products entering the Krebs cycle and resulting in physiological and structural changes in the organs. Metabolic blockage such as ketogenic diet broken up by b -oxidation and producing acetyl-CoA can improve the patients. Sirtuins play a role in the production of acetyl-CoA through oxidation of fatty acids and other pathways. Thus, we hypothesize that the targets and bioactive compounds targeting mitochondrial sirtuins can be involved in the treatment of PDC deficiency. In general, this review discusses the present knowledge on how mitochondrial sirtuins are involved in the regulation of PDC as well as their possible roles in the treatment of PDC deficiency.
Introduction
The human pyruvate dehydrogenase (PD) is a multi-enzyme complex that is involved in the production of nicotinamide adenine dinucleotide dehydrogenase (NADH), carbon dioxide (CO2), and acetyl-coenzyme A (CoA) by pyruvate decarboxylation. The common pyruvate dehydrogenase complex (PDC) has three main enzymes and five coenzymes7, 6, 5, 4, 3, 2, 1. The three components enzymes PDC include pyruvate dehydrogenase (E1, a heterotetramer of two types, with EC 1.2.4.1), dihydrolipoamide acetyltransferase (E2, EC 2.3.1.12) and dihydrolipoamide dehydrogenase (E3, EC 1.8.1.4,)9, 8. E3 binding protein (E3BP) is an additional subunit that exists in humans, and integrates stable E3 into the E2 core of each assembly11, 10, 9. Each assembly consists of five coenzymes, including thiamine pyrophosphat, lipoic acid, CoA, flavin adenine dinucleotide, and nicotinamide adenine dinucleotide (NAD+). PDC and its regulatory enzymes have an important role in all mitochondria of higher eukaryotes. This complex is active in aerobic conditions that require glucose decomposition to produce energy, and is suppressed when glucose is in short supply. In general, this enzyme is regulated in two different ways, end-product inhibition by acetyl CoA and NADH12 and phosphorylation and dephosphorylation subunit E1, which are a non-phosphorylated active form and a phosphorylated inactive form. However, its regulation is not fully understood. PDC deficiency is a mitochondrial defect, whose gene is encoded in the nucleus and the most identified motive over neonatal encephalopathies with primary lactic acidosis13. PDC deficiency may be triggered by the defects of E1α, E1β, E2, or E3 subunits. This deficiency is often due to alteration in the E1 gene located on X chromosome; all other causes are due to defects in recessive genes16, 15, 14. Several strategies have been reported to be useful in the treatment of PDC deficiency. These encompass the use of ketogenic diets (KD), administration of dichloroacetate and thiamine supplements. Unfortunately, none of these is generalized16, 15, 14 .


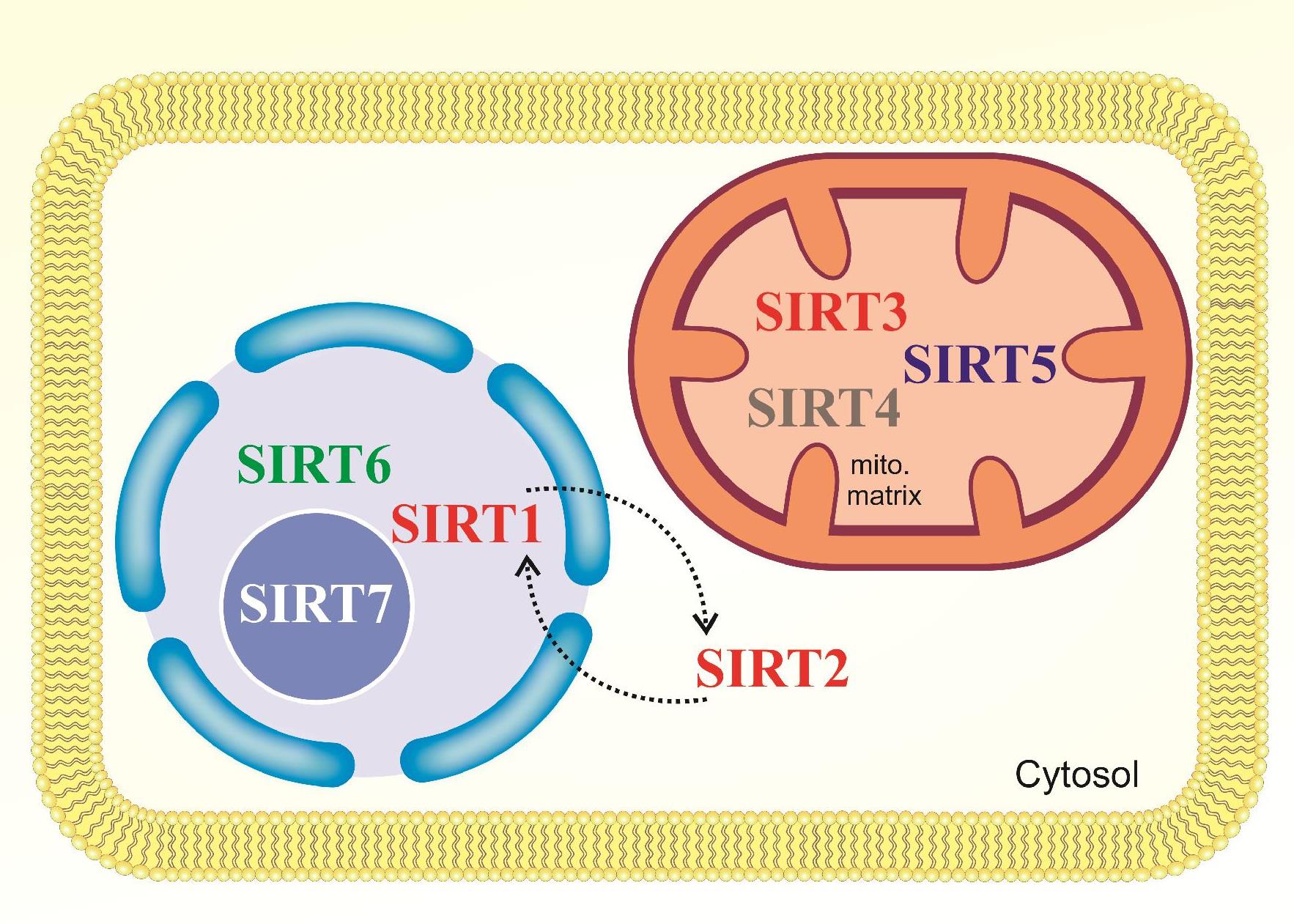
Sirtuins are protected enzymes in most species, including humans and bacteria. They rely on NAD+ for their reaction (Figure 1). Recently, it has been observed that sirtuins can catalyze reactions including deacetylation, adenosine diphosphate (ADP) ribosylation, and deacylation17. The mammalian deacetylases are divided into four classes I, II, III, and IV, based on sequence homology18. Classes I, II, and IV mammalian deacetylases include enzymes that contain zinc as a cofactor in their structure. The zinc cofactor plays a role in activating water molecules as a nucleophile and hydrolysis of the acetamide bond in acetate and lysine ɛ-amino group19. Class III mammalian deacetylase proteins, which are called sirtuins, have zinc molecules in their structure; however, the zinc elements are not directly involved in the reaction. Instead, Sirtuins utilize nicotinamide adenine dinucleotide (NAD+) cosubstrate as a cofactor to produce ADP-ribose as a nucleophile20. ADP-ribose remains for ADP-ribosylation of the substrate, alternatively on deacetylation, but this reaction is likely to stay aside response21. In mammals, seven sirtuins have been discovered, ranging from Sirt1 to Sirt7. Sirt1 and Sirt2 are present both in the nucleus and in the cytoplasm, while Sirt6 and Sirt7 are only seen in the nucleus. Sirt3, Sirt4, and Sirt5 are known as mitochondrial Sirtuins (Figure 2)22. Based on phylogenetic conservation of the core domain, sirtuins are categorized into five subclasses (I–IV and U). Subclass I contains Sirt1, Sirt2, and Sirt3 with deacetylase activity. Subclass II sirtuins include Sirt4, which exhibits ADP-ribosyltransferase activity24, 23. Subclass III sirtuins include Sirt5 that exhibits deacylase activity25 and weak deacetylase activity26. Subclass IV sirtuins contains Sirt6 and Sirt7 that have deacetylase and ADP ribosyltransferase activity27. Subclass U sirtuins are exhibited in archaea and bacteria and are intermediate between classes I and IV28.
Among the three sirtuins present in the mitochondria matrix (Sirt3, Sirt4, and Sirt5), Sirt3 is the major mitochondrial deacetylase and performs an important control for energy metabolism30, 29. On the other hand, Sirt4 is a regulator fuel used in the mitochondria32, 31. However, the role of Sirt4 in the regulation of mitochondrial metabolic pathways is unknown. Sirt5 is another type of mitochondrial sirtuins that has a role in demalonylase and desuccinylase activities in mammals. These sirtuins play an important role in metabolic, physiological, and biological processes, generally through posttranslational modifications34, 33. Posttranslational modifications are involved in the regulation of PDC, but their regulation is not well-defined. Considering the comprehensive role of sirtuins in post-translational modifications and the regulation of metabolic processes, this review was conducted to determine whether sirtuins are involved in the regulation of PDC or not. Deficiency of PDC causes pyruvate to be converted into lactate and alanine instead of acetyl-CoA because this enzyme is in the gateway of complete oxidation of glucose products entering the Krebs cycle. Metabolic blockage such as ketogenic diet is broken up by β-oxidation, and production of acetyl-CoA can improve the condition. The therapies used for the treatment of this deficiency have not been fully successful. Sirtuins are involved in the production of acetyl-CoA through oxidation of fatty acids and other pathway complexes. Thus, pharmacological targets and bioactive compounds targeting mitochondrial sirtuins can be involved in the treatment of PDC deficiency. This review discusses the present knowledge on how mitochondrial sirtuins is involved in the regulation of PDC as well as their possible roles in the treatment of PDC deficiency.
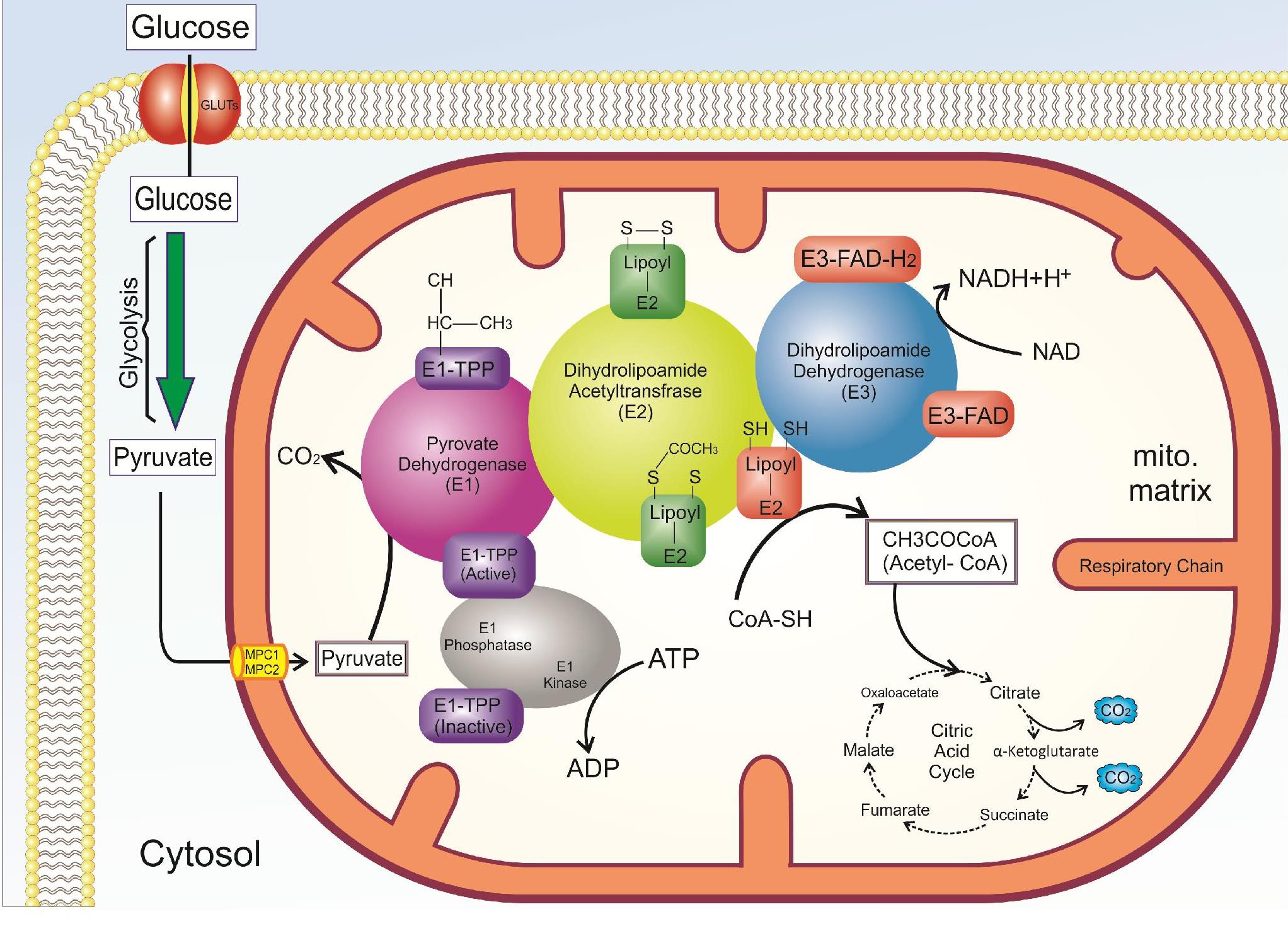

PDC and its regulatory factors
PDC catalyzes the oxidative decarboxylation reaction of pyruvate to acetyl-CoA. The function of this enzyme is irreversible and has a regulatory role in the entry of glucose products into mitochondria. This enzyme interlinks glycolysis to the Krebs cycle, and subsequently to the respiratory chain, therefore, it can be an important for the synthesis of adenosine triphosphate (ATP) by the transfer of electrons (NADH, FADH) to the respiratory chain35. In lipogenic tissues, PDC can produce fatty acids and cholesterol through the production of acetyl-CoA. PDC plays an important role in the choice of fuel and summarized metabolic regulation, which is called the “glucose-fatty acid cycle”36. This term shows the relationship between the use of glucose or fatty acid in muscle cells. The PDC of each assembly includes three component enzymes termed pyruvate dehydrogenase (PD) (E1), dihydrolipoamide acetyltransferase (E2), and dihydrolipoamide dehydrogenase (E3)9, 8. Human PDC also has an additional subunit called E3BP that integrates stable E3 into the E2 core of each assembly11, 10, 9. PDC-E1 plays a role in the oxidative decarboxylation of pyruvate and subsequently causes the production and transfer coenzyme A in the form of acetyl-CoA to cellular metabolism. To do this, PDC-E1 needs thiamine38, 37. PDC-E2 is another component that plays an essential role in the compartment and function of this multi-enzyme complex39, which needs lipoamide in its function. PDC-E3 is involved in electron transfer reactions between flavin adenine dinucleotide (FAD+) and NAD+ (Figure 3). There are several copies of these three catalytic enzymes in the PDC. In addition to these enzymes, there are two other enzymes called pyruvate dehydrogenase kinase (PDK, EC 2.7.1.99) and pyruvate dehydrogenase phosphatase (PDP, EC 3.1.3.43) that interact with PDC complex and have a regulatory role (Figure 4)11, 10, 9. In general, this enzyme is regulated in two different ways including end-product inhibition by acetyl CoA and NADH12, and phosphorylation and dephosphorylation subunit E1, which are the non-phosphorylated active form and phosphorylated inactive form40. Nevertheless, PDC is mostly regulated by kinase and phosphatase41, both of which, the control mechanisms often depend on the same metabolic effectors42. More substantial physiological roles occur via phosphorylation and dephosphorylation of three serine residues located on the α-subunit (E1) catalyzed by PDK and PDP. In mammalian tissues, there are four isoenzymes for PDKs and two isoenzymes for PDP43. The basis of pyruvate kinase is apparently unrelated tyrosine kinases and serine-threonine kinases in other mammalian regulatory processes44, 43. Although PDK is associated with the kinase family, the regulation of PDC activity by PDK results from diverse allosteric modulators. To increase the NADH ratio to NAD+, and acetyl-CoA to CoASH, increases the activity of allosteric pyruvate kinase, and results in the inhibition of PDC activity in response to a lack of mitochondrial demand47, 46, 45, 6, 4, 3. Elevated pyruvate and ADP levels inhibit the activity of PDK (activating PDC). In general, PDC is activated through its substrates, CoA-SH and NAD+, and kinase inhibition, and is inhibited by its products, acetyl CoA and NADH, and activation of the kinase. In addition, Mg2+ and Ca2+ regulate the catalytic activity of PDP, which increases the activity of this enzyme 10 times in the presence of calcium. PDP is inactivated in the absence of Ca2+ and Mg2+48, 46, 45, 44, 43, 42, 41, 40, 39, 38, 37, 36, 33, 32, 31, 30, 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6. The enzymatic activity of PDP relies on the tissue type and the mitochondrial matrix concentrations of Ca2+, Mg2+, NADH, and insulin49.
PDC deficiency
Mutation in the gene of PDC enzyme is well-known. PDC deficiency occurs due to mutations in the coding genes of E1α, E1β, E2, or E3 subunits . The mutation in the pyruvate dehydrogenase E1 (PDH)A1 gene, which encodes the alpha subunit of E1, is the most common mutation50, but this mutation is rare in the genes encoding subunits E1 β (PDHB)51, E2 (DLAT)51, E3 (DLD)52, and E3-binding protein (PDHX)53. The mutation in the gene encoding the subunit E1 is located on X chromosome and the defect observed in other genes of the subunits is recessive16, 15, 14. The mutation in the X chromosome is X-linked dominant when both females and males have the same defect in the E1 gene. In 25% children with PDHE1α deficiency, their mothers carry this mutation. In the rest of most cases, the deficiency of PD is due to new mutations in the E1α, and in general, its recurrence is low within any one of the same family54. The PDHB, which encodes the E1β subunit of PDC, contains a small amount of PDC deficiency. Although the severity of the symptoms observed in PDHB is similar to that of the PDHA1, it presents considerable phenotypic variability and severity. Defects in other sub-types such as PDP are rare and lead to acidosis in the neonatal period55, 51.
Sperl et al.56 showed that 75.5% of deficiency of PD in their patients included the mutations in PDHE-1α, which showed the same symptoms, and another 13.5% was due to deficiency of thiamin cofactor. Therefore, an important reason of PDC deficiency is X-linked. More than 120 different mutations have been detected in the E1α subunit of PDC59, 58, 57, 51. PDC deficiency resulted from E3 deficiency is due to a defect in several α-ketoacid dehydrogenases. In general, the activity of PDC is low in these individuals, which shows early clinical onset, and usually, these people die during childhood. Because PDC deficiency causes pyruvate to be converted into lactate and alanine rather than to acetyl-CoA, this enzyme is in the gateway of complete oxidation of glucose products before entering the Krebs cycle. The conversion of glucose into lactate reduces the oxidation of glucose and reduces the amount of ATP generated by the cyclic electron transport and respiratory chain. PDC deficiency causes aggravated hyperpyruvicaemia, lactic acidaemia, and hyperalaninemia60.
Clinical signs of congenital PDC deficiency
The mutation in the PDHA1 gene, which encodes the alpha subunit of E1, is the most common mutation50. Symptoms that are commonly seen in PDHE1α deficiency include delayed growth, hypotonia, ataxia, and seizures. In hemizygous males, the three main symptoms are lactic acidosis during neonatal, Leigh’s encephalopathy (the most important respiratory distress or episodic weakness), and periodic ataxia. Clinical symptoms in females with PDHE1α deficiency are usually uniform although the severity of the incidence depends on variable lyonization61. These symptoms include microcephaly, severe to moderate mental retardation, dysmorphic features, and spastic di- or quadriplegia. Deficiency of PDHE1β has been reported in a few cases64, 63, 62, 57. The cause of this deficiency is an increase in proteasome-mediated degradation, and a role of this proteasome is destroying ubiquitinated E1β subunits62. Symptoms that are commonly seen in PDHE1α deficiency include lactic acidosis (early-onset), severely delayed growth with slowly progressive neurological features of the brain stem, basal ganglia, and Leigh syndrome34. The most common features of E3BP deficiency (formerly protein X) are delayed psychomotor progress, hypotonia, and long-term survival67, 66, 65, 56. Neonatal lactic acidosis slowly progresses with thin corpus callosum and subependymal cysts. Seven cases of E1-phosphatase deficiency were found with hypotonia, feeding trouble, delayed psychomotor progress, and a lethal phenotype in infantile. Mutations in the PDK isoenzyme 3 (PDK3) gene have been found in Charcot-Marie-Tooth disease type 6. PDK3 mutates through hyperphosphorylation of the PDC, leading to peripheral neuropathy69, 68.
The genetic defect in the PDC further causes a defect in the nervous system. However, it can lead to abnormality in the function of other tissues such as muscle and liver. In the brain and other tissues, glucose normally generates ATP after oxidation in the glycolysis and TCA cycle. Consequently, PDC deficiency reduces the amount of ATP in the brain and other organs. In addition to the decreased ATP in the brain, the amount of neurotransmitter acetylcholine production decreases and causes the symptoms to be observed in the disease70. The history and Biochemical concomitants of congenital PDC deficiency, such as mental retardation, peripheral neuropathy, hypotonia, ataxia, and exercise intolerance in children, are significantly similar to the disease observed during aging71. No proven cure exists for the congenital deficiency of PDC and most patients die in the first two decades of life.

Mitochondrial Sirtuins
Sirt3, Sirt4, and Sirt5 with targeting sequences in their N-terminal, which are found in the mitochondrial matrix, are known as mitochondrial sirtuins. Among the three sirtuins present in the mitochondria matrix, Sirt3 is the major mitochondrial deacetylase, which perform an important control for energy metabolism29. For this reason, Sirt3 is highly expressed in many active tissues, such as heart, brain, kidney, liver, brown adipose tissue and muscle72. Sirt3 is initially synthesized as an inactive enzyme protein and enters the mitochondrial matrix because of the targeting sequence in N-terminal. After the translocation of this protein, the mitochondrial matrix cleavage in the mitochondria by peptidase73, which results in the processed 142 amino acids from the N terminal and subsequently activates it75, 74.
Sirt4, which is localized in the mitochondria, is a regulator fuel used in the mitochondria32, 31 and is highly expressed in tissues such as pancreatic beta cells, liver, heart, brain, and kidney32, 24, 22. Sirt4 have mono ADP ribosyltransferase and lipoamidase activity (Figure 5)32. Sirt4 is involved in regulating energy consumption and metabolic processes in the mitochondria32, 31. However, its complete mechanism is unknown.

Sirt5 is another type of mitochondrial sirtuins that plays a role in demalonylase and desuccinylase activities in mammals (Figure 6 )76. Sirt5 has shown poor deacetylase activity and it lacks ADP ribosyltransferase activity77. Sirt5 is able to remove acyl modifications such as succinylation, acetylation, and malonylation on lysine residues. Therefore, Sirt5 appears to be a deacylase rather than a deacetylase25.
Mitochondrial sirtuins and their regulatory role in PDC deficiency
Protein posttranslational modifications (PTMs) are indicators of key regulation of cellular processes and proteome diversity. Recently, high-resolution mass spectrometry (MS) has a great contribution to understand an array of modern PTM. In particular, Lys residues are the targets of numerous PTM. Lys groups can fall under acetylation, methylation, biotinylation, ubiquitination79, 76, 78, and other ubiquitination processes like butyrylation, crotonylation, propionylation, succinylation and malonylation81, 80, 79, 78, 76. PDC is known to be regulated through phosphorylation and dephosphorylation by specific PDK and PDP82. PDK1 inactivates PDC by phosphorylation of serine residues, especially in the S293, S300, and S232 positions of PDHA1, whereas PDP1 activates PDC through PDHA1 dephosphorylation. Phosphorylation of the E1 subunit can affect its E1 acetylation86, 85, 84, 83. Previous studies have reported that Lys acetylation in PDHA1 and PDP1 happens in various cancer cells and epidermal growth factor (EGF) stimulated cells. K321 acetylation of PDHA1 causes the employment of PDK1 to PDHA1, thereby improving PDHA1 phosphorylation. PDHA1 phosphorylation will inhibit it. On the other hand, the PDP1 acetylation inhibits the PDP1, and thereby, dissociates its substrate PDHA1. As a result, it will not be able to dephosphorylate and activate PDHA1. Both of these factors play important roles in promoting or inhibiting glycolysis in cancerous cells and tumor growth. Studies have shown that mitochondrial Sirt3 and acetyl-CoA acetyltransferase 1 (ACAT1) regulate deacetylase and acetyltransferase. ACAT1 causes Lys acetylation, and consequently, inhibits PDHA1 and PDP (In general, inhibition of PDC). However, Sirt3 activates Lys deacetylase, and consequently, PDHA1 and PDP1 (In general, activation of PDC). Since the cancer cells are dependent on glycolysis, these metabolic changes are important to them. Therefore, ACAT1 and SIRT3 regulate PDHA1 and PDP1 in the mitochondria through acetylation and deacetylation87, 84.
The transcription factor of hypoxia inhibitory factor1-α (HIF1-α) is involved in the transcription of numerous genes involved in encoding glycolysis enzymes, including metabolism and glucose transporters. It has been shown that HIF1-α reduces PDC and oxidative phosphorylation (OXPHOS) activity through transactivation of PDKs88. Overexpression of HIF1-α occurs in tissues with defective PDC. The production of oxidative stress by complex III leads to increased expression of HIF1-α in PDC deficiency89. Sirt3 leads to inhibition of HIF-1α activity by decreasing ROS production90. Sirt3 reduces the production of ROS in mitochondria and cytoplasm through deacetylation of complex III of ETC91 and inhibits HIF-1α activity. Inhibition of HIF-1α activity leads to reduced transactivation of PDKs, and as a result, increases PDC activity. Therefore, Sirt3 leads to an increase in PDC activity by inhibition of HIF-1α and transactivation of PDKs.
The lipoamide cofactors are bound to E2 transferase enzymes (Figure 2) and are required for PD activity92, forming PD catalytic core. The activity of deacetylase Sirt4 is low and most of its role is through lipoyl and biotinyllysine modifications. Conversely, Sirt3, Sirt4 regulate the PDC via the hydrolysis of the lipoamide cofactors in the E2 component. Sirt4 decreases the PDC activity by lipoamidase activity, which subsequently generates acetyl-CoA in human cell lines and mouse models87.
One of the post-translational modifications is succinylation of proteins. Succinylation occurs on Lys residues and its origin succinyl-CoA93. Succinylation can decrease or increase the activity of enzymes. For example, succinylation of PDC and succinate dehydrogenase complexes increases the complex activity94. Conversely, succinylation of 3-hydroxy-3-methylglutaryl-CoA synthetase 2 (HMGCS2) reduces the activity of the enzymes95. Sirt5 is another type of mitochondrial sirtuin that is capable to move desuccinylated Lys residues in proteins96. As a result, Sirt5 reduces the activity of PDC through desuccinylase activity.
Consequently, Sirt3 increases the activity of PDC through deacetylation of PDHA1 and PDP1, while Sirt4 decreases the PDC activity through lipoamidase activity and Sirt5 through desuccinylase activity.
Metabolic and pharmacologic management of PDC
In general, people with PDC deficiency are not very recognizable and their treatment is not adequately effective. Because PDC deficiency causes pyruvate to be converted to lactate and alanine rather than to acetyl-CoA, this enzyme is in the gateway of complete oxidation glucose products entering the Krebs cycle. The conversion of glucose to lactate reduces the oxidation of glucose and reduces the amount of ATP generated by the cyclic cycle and respiratory chain. PDC deficiency causes aggravated hyperpyruvicaemia, lactic acidaemia, and hyperalaninemia60. Several strategies have been reported to be useful in the treatment of PDC deficiency, which encompass the use of ketogenic diet, administration of dichloroacetate, and thiamine supplements97. However, none of these strategies are generalized, and they have been reported with variable success.
Metabolic management of dichloroacetate in PDC deficiency and hypotheses related to the role of sirtuins in this disease
PDC is regulated through phosphorylation and dephosphorylation by specific PDK and PDP82. PDK1 inactivates PDC by phosphorylation of serine residues, especially in S293, S300, and S232 positions of PDHA1, whereas PDP1 activates PDC through PDHA1 dephosphorylation87. The regulation of PDC activity by PDKs and PDPs results from diverse allosteric modulators. One of these allosteric modulators is pyruvate, which inhibits the activity of PDKs. Many drugs inhibit all PDK isoforms except PDK3 (testes-specific PDK3) and result in changes in the phosphorylation of the E1 subunits in PDC.
Due to the structural similarity (structural analog) between dichloroacetate (DCA) and pyruvate, DCA can be thought to: 1) Inhibit E1 kinase, and consequently PDC phosphorylation98, 2) Increase the transferring of pyruvate into mitochondria, and 3) Reduce lactate levels in the serum99, leading to increased activity of PDC. DCA is used primarily for the treatment of lactic acidemia in PDC deficiency or respiratory chain defects68, 63. It has been observed that the combination of phenylbutyrate with DCA results in increased activity of PDC in mice100. Studies have shown that phenylbutyrate in the fibroblasts of patients with PDC deficiency can also increase the PDC activity101.
Sirt3 causes dephosphorylation of PDC by deacetylation PDHA1 and PDP1, and thus, activating PDC (Lys acetylation is inhibiting PDHA1 and PDP1). On the other hand, Sirt3 decreases PDK activity by reducing HIF-1α and leads to activation of PDC. Therefore, Sirt3 functions similarly to DCA through these two mechanisms.
Short-term consumption of DCA is beneficial for patients with PDC deficiency102. However, long-term treatment with DCA causes reversible peripheral neuropathy. For this reason, DCA treatment has remained controversial. Perhaps giving the compounds that activate Sirt3 does not have this harm.
Metabolic management of KD in PDC deficiency and hypotheses related to the role of sirtuins in this disease
Most probably, the best way to treat patients with PDC deficiency is the use of ketogenic supplements103. Consumption of KD provides the long-chain saturated triglycerides. Providing high-fat foods, instead of glucose-rich foods, reduces glycolysis and increases the production of acetyl-CoA through the beta-oxidation pathway. The increased production of acetyl-CoA due to oxidation of fatty acids leads to the production of hydroxybutyrate and acetoacetate through ketogenesis. Acetyl-CoA produced by ketone bodies provides acetyl-CoA for the Krebs cycle, which should be provided through PDC60.
Several theories have suggested that KD reduces seizures105, 104. Because ATP generation via mitochondria instead of glycolysis selectively activates the potassium channel, it has stabilizing effects on the neuronal cell membrane107, 106. KD may facilitate this reaction by increasing the production of γ-aminobutyric acid in the brain tissue. Hence, the hyper-polarization of neurons stabilizes the synaptic function and reduces seizures in the brain.
Acetoacetate, β-hydroxybutyrate, and acetone are known as ketone bodies that are made from Acetyl-CoA. Acetyl-CoA is converted to 3-hydroxy-3-methylglutaryl-CoA (HMGCoA) by methylglutaryl-CoA synthase 2 (HMGCS2), the rate-limiting step in ketone body biosynthesis. Eventually, HMGCoA is converted into ketone bodies. Sirt3 and Sirt5 activate the HMGCS2 enzyme and produce ketone bodies through deacetylation and desuccinylation of HMGCS2, respectively108. Therefore, Sirt3 and Sirt5 play a role in the treatment of PDC by producing ketone bodies. PDC deficiency inhibits the production of Acetyl-CoA by glucose. Thus, acetyl-CoA of the TCA cycle needs to be achieved through oxidation of fatty acids. Sirt3 produces acetyl-CoA during starvation by deacetylation of the long-chain acyl-CoA dehydrogenase (LCAD), activation of this key enzyme, and beta-oxidation of fatty acids109. Sirt3 also produces acetyl-CoA in extracellular tissues through deacetylation and activation of acetyl-CoA synthetase 2111, 110. As a result, Sirt3 can improve the symptoms of PDC through fatty acid catabolism in the liver and the production of acetyl-CoA from acetate in extra-liver tissues.
Sirt4 is another critical regulator of fatty acid metabolism and leads to inhibition of oxidation of fatty acids. Sirt4 causes deacetylation of malonyl CoA decarboxylase (MCoAD), thereby activating it. MCoAD plays an important role in the metabolism of fatty acids112. Studies on mice have shown that Sirt4 knock-out increases fatty acid oxidation in different tissues112, 31. Thus Sirt4, in contrast to Sirt3, is a negative regulator of fatty acid oxidation, indicating that Sirt3 and Sirt4 are mutually involved in the regulation of lipid metabolism.
Sirt5 is also involved in the regulation of fatty acid metabolism via desuccinylation. It is known that 60% of the proteins involved in the metabolism of lipids are succinylated. One of the important enzymes involved in the oxidation of fatty acids is hydroxyl coenzyme A dehydrogenase, which is regulated by succinylation and desuccinylation. Sirt5 activates this enzyme via desuccinylation94. The overall consequence is that Sirt3 and Sirt5 improve the condition of the disease by activating the key enzymes of the ketone body pathway and fatty acid oxidation, resulting in increased production of acetyl-CoA. However, Sirt4 works the opposite.
Other hypotheses related to the role of sirtuins in the treatment of PDC deficiency
Glucose is converted to pyruvate and lactic acid via the glycolysis pathway. The production of lactic acid increases the PDC deficiency. As a result, reducing glycolysis in PDC is desirable and reduces the amount of lactic acid production. Sirt3 can reduce the coupling of hexokinase to the voltage-dependent ion channels through cyclophilin D deacetylation, and thus the mitochondria membrane113. Preventing hexokinase II binding to the mitochondrial membrane reduces glucose-6-phosphate production, and thus, the glycolysis pathway. Pyruvate kinase M1/M2 is another key enzyme of the glycolysis pathway, which is regulated by succinylation. Sirt5 is involved in the glycolysis pathway regulation through the desuccinylation of K498 Lys residue of this enzyme94. Consequently, Sirt3 and Sirt5 are regulated by the glycolysis pathway and can serve as important therapeutic targets.
Conclusion and future perspectives
PD is a multi-enzymatic mitochondrial complex that converts pyruvate into acetyl-CoA, CO2, and NADH. This complex consists of three main enzymes (pyruvate dehydrogenase, dihydrolipoamide acetyltransferase, and dihydrolipoamide dehydrogenase) and two regulatory enzymes (PDK and PDP). In general, this complex is regulated in two different ways including end-product inhibition12 and posttranslational modifications (phosphorylation and dephosphorylation of subunit E1). Phosphorylation is catalyzed by PDK and dephosphorylation is catalyzed by PDP. The regulation of PDC activity by PDK and PDP results in diverse allosteric modulators such as NAD+, NADH, CoASH, acetyl-CoA, pyruvate, ADP, Ca2+, and Mg2+47, 46, 49, 45, 6, 4, 3. Sirtuins are the class III of histone deacylatases that catalyze protein posttranslational modifications, including deacetylation, ADP ribosylation, and deacylation. Sirt3, Sirt4, and Sirt5 are mitochondrial sirtuins that control PDC by allosteric regulation. Sirt3 enhances the activity of PDC through deacetylation of PDHA1 and PDP1 and inhibition of HIF-1α87, 84. Sirt4, which regulates the PDC via lipoamidase, hydrolyzes the lipoamide cofactors in the E2 component of DLAT and decreases PDC activity. Succinylation of PDC increases the complex activity94. Sirt5 is another type of mitochondrial sirtuins that causes desuccinylation of Lys residues in proteins96. It reduces the activity of this enzyme through desuccinylation of PDC. Consequently, Sirt3 increases the activity of PDC by deacetylation of PDHA1 and PDP1, while Sirt4 and Sirt5 inhibit PDC activity via lipoamidase and desuccinylase activity, respectively.
Mutation in the genes of the PDC enzyme includes a mutation in the PDHA1 gene encoding the alpha subunit of E1, DLAT gene encoding E2114, DLD gene encoding E352, and PDHX gene encoding E3-binding protein53. In the PDC deficiency, the acetyl-CoA production decreases for the Krebs cycle. In the treatment of PDC deficiency, the use of KD, administration of dichloroacetate and thiamine supplements are recommended to activate PDC and produce acetyl-CoA. Sirt3 enhances the activity of PDC through deacetylation of PDHA1, PDP1, and inhibition of HIF-1α, which appears to be similar to the action of dichloroacetate. Sirt3 can improve the symptoms of PDC by fatty acid catabolism in the liver and the production of acetyl-CoA from acetate in extra-liver tissues. Desuccinylation of β-oxidation enzymes by Sirt594 increases the production of acetyl-CoA. The overall consequence is that Sirt3 and Sirt5 improve the condition of the disease by activating the key enzyme of the ketone body pathway and fatty acid oxidation, and elevate the production of acetyl-CoA, while Sirt4 works in the opposite manner. In conclusion, mitochondrial sirtuins are involved in regulating the activity and treatment of PDC deficiency. The compounds can possibly regulate mitochondrial sirtuins, which can be involved in the regulation of PDC. However, extensive studies are required to discover the role of sirtuins in the regulation of PDC.
ABBREVIATIONS
ACAT1: Acetyl-CoA acetyltransferase 1
ADP: Adenosine diphosphate
ATP: Adenosine triphosphate
CO2: Carbon dioxide
CoA: Coenzyme A
DCA: Dichloroacetate
FAD+: flavin adenine dinucleotide
HIF-1α: Hypoxia inhibitory factor1-α
HMGCoA: 3-hydroxy-3-methylglutaryl-CoA
HMGCS2: 3-hydroxy-3-methylglutaryl-CoA synthetase 2
KD: Ketogenic diet
MCoAD: Malonyl CoA decarboxylase
NAD+: Nicotinamide adenine dinucleotide
NADH: Nicotinamide adenine dinucleotide dehydrogenase
PD: Pyruvate dehydrogenase
PDC: Pyruvate dehydrogenase complex
PDH: Pyruvate dehydrogenase E1
PDK: Pyruvate dehydrogenase kinase
PDP: Pyruvate dehydrogenase phosphatase
PTM: Protein posttranslational modifications
Sirt: Sirtuin
Competing Interests
The authors declare that they have no conflicts of interest.
Authors' Contributions
AN and RK contributed to the design of the research. AN and SK extracted the data and summarized it. AV-R and RK edited the first draft. MS and ZA reviewed the first draft. All authors approved the final draft.
Acknowledgements
The financial supports of Kermanshah University of Medical Sciences are gratefully acknowledged (grant number: 97325).
References
-
Behal
R.H.,
Buxton
D.B.,
Robertson
J.G.,
Olson
M.S.,
Regulation of the pyruvate dehydrogenase multienzyme complex. Annu Rev Nutr.
1993;
13
(1)
:
497-520
.
View Article PubMed Google Scholar -
Frey
P.A.,
Flournoy
D.S.,
Gruys
K.,
Yang
Y.S.,
Intermediates in reductive transacetylation catalyzed by pyruvate dehydrogenase complex. Ann N Y Acad Sci.
1989;
573
(1)
:
21-35
.
View Article PubMed Google Scholar -
Patel
M.,
Korotchkina
L.,
Regulation of the pyruvate dehydrogenase complexPortland Press Limited 2006.
View Article Google Scholar -
Patel
M.S.,
Roche
T.E.,
Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J.
1990;
4
(14)
:
3224-33
.
View Article PubMed Google Scholar -
Perham
R.N.,
Packman
L.C.,
2-Oxo acid dehydrogenase multienzyme complexes: domains, dynamics, and design. Ann N Y Acad Sci.
1989;
573
(1)
:
1-20
.
View Article PubMed Google Scholar -
Roche
T.E.,
Hiromasa
Y.,
Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol Life Sci.
2007;
64
(7-8)
:
830-49
.
View Article PubMed Google Scholar -
Wieland
O.H.,
The mammalian pyruvate dehydrogenase complex: structure and regulation. Reviews of Physiology, Biochemistry and PharmacologySpringer 1983.
Google Scholar -
Smolle
M.,
Prior
A.E.,
Brown
A.E.,
Cooper
A.,
Byron
O.,
Lindsay
J.G.,
A new level of architectural complexity in the human pyruvate dehydrogenase complex. J Biol Chem.
2006;
281
(28)
:
19772-80
.
View Article PubMed Google Scholar -
Zhou
Z.H.,
McCarthy
D.B.,
O'Connor
C.M.,
Reed
L.J.,
Stoops
J.K.,
The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA.
2001;
98
(26)
:
14802-7
.
View Article PubMed Google Scholar -
Maj
M.C.,
Cameron
J.M.,
Robinson
B.H.,
Pyruvate dehydrogenase phosphatase deficiency: orphan disease or an under-diagnosed condition?. Mol Cell Endocrinol.
2006;
249
(1-2)
:
1-9
.
View Article PubMed Google Scholar -
Yu
X.,
Hiromasa
Y.,
Tsen
H.,
Stoops
J.K.,
Roche
T.E.,
Zhou
Z.H.,
Structures of the human pyruvate dehydrogenase complex cores: a highly conserved catalytic center with flexible N-terminal domains. Structure.
2008;
16
(1)
:
104-14
.
View Article PubMed Google Scholar -
Butterworth
R.F.,
Neurotransmitter function in thiamine-deficiency encephalopathy. Neurochem Int.
1982;
4
(6)
:
449-64
.
View Article PubMed Google Scholar -
Huang
B.,
Gudi
R.,
Wu
P.,
Harris
R.A.,
Hamilton
J.,
Popov
K.M.,
Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J Biol Chem.
1998;
273
(28)
:
17680-8
.
View Article PubMed Google Scholar -
Brown
G.K.,
Otero
L.J.,
LeGris
M.,
Brown
R.M.,
Pyruvate dehydrogenase deficiency. J Med Genet.
1994;
31
(11)
:
875-9
.
View Article PubMed Google Scholar -
Meirleir
L. De,
Defects of pyruvate metabolism and the Krebs cycle. J of chil neu.
2002;
17
(3S)
:
26-33
.
-
Hansen
L.L.,
Brown
G.K.,
Kirby
D.M.,
Dahl
H.H.,
Characterization of the mutations in three patients with pyruvate dehydrogenase E1 α deficiency. J Inherit Metab Dis.
1991;
14
(2)
:
140-51
.
View Article PubMed Google Scholar -
Imai
S.,
Armstrong
C.M.,
Kaeberlein
M.,
Guarente
L.,
Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature.
2000;
403
(6771)
:
795-800
.
View Article PubMed Google Scholar -
Xu
W.S.,
Parmigiani
R.B.,
Marks
P.A.,
Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene.
2007;
26
(37)
:
5541-52
.
View Article PubMed Google Scholar -
Holbert
M.A.,
Marmorstein
R.,
Structure and activity of enzymes that remove histone modifications. Curr Opin Struct Biol.
2005;
15
(6)
:
673-80
.
View Article PubMed Google Scholar -
Sauve
A.A.,
Wolberger
C.,
Schramm
V.L.,
Boeke
J.D.,
The biochemistry of sirtuins. Annu Rev Biochem.
2006;
75
(1)
:
435-65
.
View Article PubMed Google Scholar -
Du
J.,
Jiang
H.,
Lin
H.,
Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD. Biochemistry.
2009;
48
(13)
:
2878-90
.
View Article PubMed Google Scholar -
Michishita
E.,
Park
J.Y.,
Burneskis
J.M.,
Barrett
J.C.,
Horikawa
I.,
Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell.
2005;
16
(10)
:
4623-35
.
View Article PubMed Google Scholar -
Haigis
M.C.,
Guarente
L.P.,
Mammalian sirtuins emerging roles in physiology, aging, and calorie restriction. Genes Dev.
2006;
20
(21)
:
2913-21
.
View Article PubMed Google Scholar -
Ahuja
N.,
Schwer
B.,
Carobbio
S.,
Waltregny
D.,
North
B.J.,
Castronovo
V.,
Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem.
2007;
282
(46)
:
33583-92
.
View Article PubMed Google Scholar -
Yu
J.,
Sadhukhan
S.,
Noriega
L.G.,
Moullan
N.,
He
B.,
Weiss
R.S.,
Metabolic characterization of a Sirt5 deficient mouse model. Sci Rep.
2013;
3
(1)
:
2806
.
View Article PubMed Google Scholar -
Nakagawa
T.,
Lomb
D.J.,
Haigis
M.C.,
Guarente
L.,
SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell.
2009;
137
(3)
:
560-70
.
View Article PubMed Google Scholar -
Kawahara
T.L.,
Michishita
E.,
Adler
A.S.,
Damian
M.,
Berber
E.,
Lin
M.,
SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell.
2009;
136
(1)
:
62-74
.
View Article PubMed Google Scholar -
Frye
R.A.,
Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun.
2000;
273
(2)
:
793-8
.
View Article PubMed Google Scholar -
Lombard
D.B.,
Alt
F.W.,
Cheng
H.L.,
Bunkenborg
J.,
Streeper
R.S.,
Mostoslavsky
R.,
Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol.
2007;
27
(24)
:
8807-14
.
View Article PubMed Google Scholar -
Liu
L.,
Nam
M.,
Fan
W.,
Akie
T.E.,
Hoaglin
D.C.,
Gao
G.,
Nutrient sensing by the mitochondrial transcription machinery dictates oxidative phosphorylation. The J of clini invest.
2014;
124
(2)
:
768
.
View Article Google Scholar -
Nasrin
N.,
Wu
X.,
Fortier
E.,
Feng
Y.,
Bare'
O.C.,
Chen
S.,
SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J Biol Chem.
2010;
285
(42)
:
31995-2002
.
View Article PubMed Google Scholar -
Haigis
M.C.,
Mostoslavsky
R.,
Haigis
K.M.,
Fahie
K.,
Christodoulou
D.C.,
Murphy
A.J.,
SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic β cells. Cell.
2006;
126
(5)
:
941-54
.
View Article PubMed Google Scholar -
German
N.J.,
Haigis
M.C.,
Sirtuins and the metabolic hurdles in cancer. Curr Biol.
2015;
25
(13)
:
569-83
.
View Article PubMed Google Scholar -
Sebastián
C.,
Satterstrom
F.K.,
Haigis
M.C.,
Mostoslavsky
R.,
From sirtuin biology to human diseases: an update. J Biol Chem.
2012;
287
(51)
:
42444-52
.
View Article PubMed Google Scholar -
Margineantu
D.H.,
Brown
R.M.,
Brown
G.K.,
Marcus
A.H.,
Capaldi
R.A.,
Heterogeneous distribution of pyruvate dehydrogenase in the matrix of mitochondria. Mitochondrion.
2002;
1
(4)
:
327-38
.
View Article PubMed Google Scholar -
Robinson
B.H.,
MacKay
N.,
Chun
K.,
Ling
M.,
Disorders of pyruvate carboxylase and the pyruvate dehydrogenase complex. J Inherit Metab Dis.
1996;
19
(4)
:
452-62
.
View Article PubMed Google Scholar -
Arjunan
P.,
Nemeria
N.,
Brunskill
A.,
Chandrasekhar
K.,
Sax
M.,
Yan
Y.,
Structure of the pyruvate dehydrogenase multienzyme complex E1 component from Escherichia coli at 1.85 A resolution. Biochemistry.
2002;
41
(16)
:
5213-21
.
View Article PubMed Google Scholar -
Chandrasekhar
K.,
Arjunan
P.,
Sax
M.,
Nemeria
N.,
Jordan
F.,
Furey
W.,
Active-site changes in the pyruvate dehydrogenase multienzyme complex E1 apoenzyme component from Escherichia coli observed at 2.32 A resolution. Acta Crystallogr D Biol Crystallogr.
2006;
62
(Pt 11)
:
1382-6
.
View Article PubMed Google Scholar -
Packman
L.C.,
Green
B.,
Perham
R.N.,
Lipoylation of the E2 components of the 2-oxo acid dehydrogenase multienzyme complexes of Escherichia coli. Biochem J.
1991;
277
(Pt 1)
:
153-8
.
View Article PubMed Google Scholar -
Browning
M.,
Baudry
M.,
Bennett
W.F.,
Lynch
G.,
Phosphorylation-mediated changes in pyruvate dehydrogenase activity influence pyruvate-supported calcium accumulation by brain mitochondria. J Neurochem.
1981;
36
(6)
:
1932-40
.
View Article PubMed Google Scholar -
Morgan
D.G.,
Routtenberg
A.,
Brain pyruvate dehydrogenase: phosphorylation and enzyme activity altered by a training experience. Science.
1981;
214
(4519)
:
470-1
.
View Article PubMed Google Scholar -
Garland
P.B.,
Newsholme
E.A.,
Randle
P.J.,
Regulation of glucose uptake by muscle. 9. Effects of fatty acids and ketone bodies, and of alloxan-diabetes and starvation, on pyruvate metabolism and on lactate-pyruvate and L-glycerol 3-phosphate-dihydroxyacetone phosphate concentration ratios in rat heart and rat diaphragm muscles. Biochem J.
1964;
93
(3)
:
665-78
.
View Article PubMed Google Scholar -
Sugden
P.H.,
Simister
N.E.,
Role of multisite phosphorylation in the regulation of ox kidney pyruvate dehydrogenase complex. FEBS Lett.
1980;
111
(2)
:
299-302
.
View Article PubMed Google Scholar -
Kerbey
A.L.,
Randle
P.J.,
Role of multi-site phosphorylation in regulation of pig heart pyruvate dehydrogenase phosphatase. FEBS Lett.
1979;
108
(2)
:
485-8
.
View Article PubMed Google Scholar -
Baker
J.C.,
Yan
X.,
Peng
T.,
Kasten
S.,
Roche
T.E.,
Marked differences between two isoforms of human pyruvate dehydrogenase kinase. J Biol Chem.
2000;
275
(21)
:
15773-81
.
View Article PubMed Google Scholar -
Randle
P.J.,
Fuel selection in animalsPortland Press Limited 1986.
View Article Google Scholar -
Roche
T.E.,
Baker
J.C.,
Yan
X.,
Hiromasa
Y.,
Gong
X.,
Peng
T.,
Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Res Mol Biol.
2001;
70
:
33-75
.
View Article PubMed Google Scholar -
Hansford
R.G.,
Studies on the effects of coenzyme A-SH: acetyl coenzyme A, nicotinamide adenine dinucleotide: reduced nicotinamide adenine dinucleotide, and adenosine diphosphate: adenosine triphosphate ratios on the interconversion of active and inactive pyruvate dehydrogenase in isolated rat heart mitochondria. J Biol Chem.
1976;
251
(18)
:
5483-9
.
PubMed Google Scholar -
Pratt
M.L.,
Roche
T.E.,
Mechanism of pyruvate inhibition of kidney pyruvate dehydrogenasea kinase and synergistic inhibition by pyruvate and ADP. J Biol Chem.
1979;
254
(15)
:
7191-6
.
PubMed Google Scholar -
Kanzaki
T.,
Hayakawa
T.,
Hamada
M.,
Fukuyoshi
Y.,
Koike
M.,
Mammalian α-keto acid dehydrogenase complexes. IV. Substrate specificities and kinetic properties of the pig heart pyruvate and 2-oxyoglutarate dehydrogenase complexes. J Biol Chem.
1969;
244
(5)
:
1183-7
.
PubMed Google Scholar -
Lissens
W.,
De Meirleir
L.,
Seneca
S.,
Liebaers
I.,
Brown
G.K.,
Brown
R.M.,
Mutations in the X-linked pyruvate dehydrogenase (E1) α subunit gene (PDHA1) in patients with a pyruvate dehydrogenase complex deficiency. Hum Mutat.
2000;
15
(3)
:
209-19
.
View Article PubMed Google Scholar -
Head
R.A.,
Brown
R.M.,
Zolkipli
Z.,
Shahdadpuri
R.,
King
M.D.,
Clayton
P.T.,
Clinical and genetic spectrum of pyruvate dehydrogenase deficiency: dihydrolipoamide acetyltransferase (E2) deficiency. Ann Neurol.
2005;
58
(2)
:
234-41
.
View Article PubMed Google Scholar -
Liu
T.C.,
Kim
H.,
Arizmendi
C.,
Kitano
A.,
Patel
M.S.,
Identification of two missense mutations in a dihydrolipoamide dehydrogenase-deficient patient. Proc Natl Acad Sci USA.
1993;
90
(11)
:
5186-90
.
View Article PubMed Google Scholar -
Kerr
D.S.,
Wexler
I.D.,
Zinn
A.B.,
Disorders of pyruvate metabolism and the tricarboxylic acid cycle. Inborn Metabolic DiseasesSpringer 2000.
Google Scholar -
Brown
R.M.,
Dahl
H.H.,
Brown
G.K.,
X-chromosome localization of the functional gene for the E1 α subunit of the human pyruvate dehydrogenase complex. Genomics.
1989;
4
(2)
:
174-81
.
View Article PubMed Google Scholar -
Sperl
W.,
Fleuren
L.,
Freisinger
P.,
Haack
T.B.,
Ribes
A.,
Feichtinger
R.G.,
The spectrum of pyruvate oxidation defects in the diagnosis of mitochondrial disorders. J Inherit Metab Dis.
2015;
38
(3)
:
391-403
.
View Article PubMed Google Scholar -
Imbard
A.,
Boutron
A.,
Vequaud
C.,
Zater
M.,
de Lonlay
P.,
de Baulny
H.O.,
Molecular characterization of 82 patients with pyruvate dehydrogenase complex deficiency. Structural implications of novel amino acid substitutions in E1 protein. Mol Genet Metab.
2011;
104
(4)
:
507-16
.
View Article PubMed Google Scholar -
Quintana
E.,
Gort
L.,
Busquets
C.,
Navarro-Sastre
A.,
Lissens
W.,
Moliner
S.,
Working Group
PDH,
Mutational study in the PDHA1 gene of 40 patients suspected of pyruvate dehydrogenase complex deficiency. Clin Genet.
2010;
77
(5)
:
474-82
.
View Article PubMed Google Scholar -
Ridout
C.K.,
Keighley
P.,
Krywawych
S.,
Brown
R.M.,
Brown
G.K.,
A putative exonic splicing enhancer in exon 7 of the PDHA1 gene affects splicing of adjacent exons. Hum Mutat.
2008;
29
(3)
:
451
.
View Article PubMed Google Scholar -
Saudubray
J.M.,
Baumgartner
M.R.,
Walter
J.H.,
Inborn metabolic diseases: diagnosis and treatmentSpringer 2016.
View Article Google Scholar -
De Meirleir
L.,
Specola
N.,
Seneca
S.,
Lissens
W.,
Pyruvate dehydrogenase E1 α deficiency in a family: different clinical presentation in two siblings. J Inherit Metab Dis.
1998;
21
(3)
:
224-6
.
View Article PubMed Google Scholar -
Han
Z.,
Zhong
L.,
Srivastava
A.,
Stacpoole
P.W.,
Pyruvate dehydrogenase complex deficiency caused by ubiquitination and proteasome-mediated degradation of the E1 subunit. J Biol Chem.
2008;
283
(1)
:
237-43
.
View Article PubMed Google Scholar -
Okajima
K.,
Korotchkina
L.G.,
Prasad
C.,
Rupar
T.,
Phillips
J.A.,
Ficicioglu
C.,
Mutations of the E1β subunit gene (PDHB) in four families with pyruvate dehydrogenase deficiency. Mol Genet Metab.
2008;
93
(4)
:
371-80
.
View Article PubMed Google Scholar -
Quintana
E.,
Mayr
J.,
Silva
M.G.,
Font
A.,
Tortoledo
M.,
Moliner
S.,
PDH E1β deficiency with novel mutations in two patients with Leigh syndrome. J Inherit Metab Dis.
2009;
32
(1)
:
339-43
.
View Article Google Scholar -
Brown
R.M.,
Head
R.A.,
Brown
G.K.,
Pyruvate dehydrogenase E3 binding protein deficiency. Hum Genet.
2002;
110
(2)
:
187-91
.
View Article PubMed Google Scholar -
Brown
R.M.,
Head
R.A.,
Morris
A.A.,
Raiman
J.A.,
Walter
J.H.,
Whitehouse
W.P.,
Pyruvate dehydrogenase E3 binding protein (protein X) deficiency. Dev Med Child Neurol.
2006;
48
(9)
:
756-60
.
View Article PubMed Google Scholar -
Schiff
M.,
Miné
M.,
Brivet
M.,
Marsac
C.,
Elmaleh-Bergés
M.,
Evrard
P.,
Leigh's disease due to a new mutation in the PDHX gene. Ann Neurol.
2006;
59
(4)
:
709-14
.
View Article PubMed Google Scholar -
Cameron
J.M.,
Maj
M.,
Levandovskiy
V.,
Barnett
C.P.,
Blaser
S.,
Mackay
N.,
Pyruvate dehydrogenase phosphatase 1 (PDP1) null mutation produces a lethal infantile phenotype. Hum Genet.
2009;
125
(3)
:
319-26
.
View Article PubMed Google Scholar -
Kennerson
M.L.,
Yiu
E.M.,
Chuang
D.T.,
Kidambi
A.,
Tso
S.C.,
Ly
C.,
A new locus for X-linked dominant Charcot-Marie-Tooth disease (CMTX6) is caused by mutations in the pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) gene. Hum Mol Genet.
2013;
22
(7)
:
1404-16
.
View Article PubMed Google Scholar -
Barnerias
C.,
Saudubray
J.,
Touati
G.,
Lonlay
P. De,
Dulac
O.,
Ponsot
G.,
Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Develop Medi Chil Neu.
2010;
52
(2)
:
e1-9
.
View Article Google Scholar -
Huq
A.H.,
Ito
M.,
Naito
E.,
Saijo
T.,
Takeda
E.,
Kuroda
Y.,
Demonstration of an unstable variant of pyruvate dehydrogenase protein (E1) in cultured fibroblasts from a patient with congenital lactic acidemia. Pediatr Res.
1991;
30
(1)
:
11-4
.
View Article PubMed Google Scholar -
Onyango
P.,
Celic
I.,
McCaffery
J.M.,
Boeke
J.D.,
Feinberg
A.P.,
SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci USA.
2002;
99
(21)
:
13653-8
.
View Article PubMed Google Scholar -
Schwer
B.,
North
B.J.,
Frye
R.A.,
Ott
M.,
Verdin
E.,
The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol.
2002;
158
(4)
:
647-57
.
View Article PubMed Google Scholar -
Scher
M.B.,
Vaquero
A.,
Reinberg
D.,
SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev.
2007;
21
(8)
:
920-8
.
View Article PubMed Google Scholar -
Palacios
O.M.,
Carmona
J.J.,
Michan
S.,
Chen
K.Y.,
Manabe
Y.,
Ward
J.L.,
Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1α in skeletal muscle. Aging (Albany NY).
2009;
1
(9)
:
771-83
.
View Article PubMed Google Scholar -
Peng
C.,
Lu
Z.,
Xie
Z.,
Cheng
Z.,
Chen
Y.,
Tan
M.,
The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics.
2011;
10
(12)
:
M111.012658
.
View Article Google Scholar -
Verdin
E.,
Hirschey
M.D.,
Finley
L.W.,
Haigis
M.C.,
Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci.
2010;
35
(12)
:
669-75
.
View Article PubMed Google Scholar -
Berger
S.L.,
The complex language of chromatin regulation during transcription. Nature.
2007;
447
(7143)
:
407-12
.
View Article PubMed Google Scholar -
Chen
Y.,
Sprung
R.,
Tang
Y.,
Ball
H.,
Sangras
B.,
Kim
S.C.,
Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics.
2007;
6
(5)
:
812-9
.
View Article PubMed Google Scholar -
Tan
M.,
Luo
H.,
Lee
S.,
Jin
F.,
Yang
J.S.,
Montellier
E.,
Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell.
2011;
146
(6)
:
1016-28
.
View Article PubMed Google Scholar -
Xie
Z.,
Dai
J.,
Dai
L.,
Tan
M.,
Cheng
Z.,
Wu
Y.,
Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics.
2012;
11
(5)
:
100-7
.
View Article PubMed Google Scholar -
Linn
T.C.,
Pettit
F.H.,
Reed
L.J.,
α-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc Natl Acad Sci USA.
1969;
62
(1)
:
234-41
.
View Article PubMed Google Scholar -
Holness
M.,
Sugden
M.,
Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylationPortland Press Limited 2003.
View Article Google Scholar -
Fan
J.,
Shan
C.,
Kang
H.B.,
Elf
S.,
Xie
J.,
Tucker
M.,
Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol Cell.
2014;
53
(4)
:
534-48
.
View Article PubMed Google Scholar -
Jing
E.,
O'Neill
B.T.,
Rardin
M.J.,
Kleinridders
A.,
Ilkeyeva
O.R.,
Ussar
S.,
Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes.
2013;
62
(10)
:
3404-17
.
View Article PubMed Google Scholar -
Wieland
O.,
Jagow-Westermann
B.,
ATP-dependent inactivation of heart muscle pyruvate dehydrogenase and reactivation by Mg(++). FEBS Lett.
1969;
3
(4)
:
271-4
.
View Article PubMed Google Scholar -
Mathias
R.A.,
Greco
T.M.,
Oberstein
A.,
Budayeva
H.G.,
Chakrabarti
R.,
Rowland
E.A.,
Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell.
2014;
159
(7)
:
1615-25
.
View Article PubMed Google Scholar -
Patel
K.P.,
O'Brien
T.W.,
Subramony
S.H.,
Shuster
J.,
Stacpoole
P.W.,
The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab.
2012;
106
(3)
:
385-94
.
View Article PubMed Google Scholar -
Simpson
N.E.,
Han
Z.,
Berendzen
K.M.,
Sweeney
C.A.,
Oca-Cossio
J.A.,
Constantinidis
I.,
Magnetic resonance spectroscopic investigation of mitochondrial fuel metabolism and energetics in cultured human fibroblasts: effects of pyruvate dehydrogenase complex deficiency and dichloroacetate. Mol Genet Metab.
2006;
89
(1-2)
:
97-105
.
View Article PubMed Google Scholar -
Finley
L.W.,
Carracedo
A.,
Lee
J.,
Souza
A.,
Egia
A.,
Zhang
J.,
SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell.
2011;
19
(3)
:
416-28
.
View Article PubMed Google Scholar -
Muller
F.L.,
Liu
Y.,
Van Remmen
H.,
Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem.
2004;
279
(47)
:
49064-73
.
View Article PubMed Google Scholar -
Rahmatullah
M.,
Radke
G.A.,
Andrews
P.C.,
Roche
T.E.,
Changes in the core of the mammalian-pyruvate dehydrogenase complex upon selective removal of the lipoyl domain from the transacetylase component but not from the protein X component. J Biol Chem.
1990;
265
(24)
:
14512-7
.
PubMed Google Scholar -
Zhang
Z.,
Tan
M.,
Xie
Z.,
Dai
L.,
Chen
Y.,
Zhao
Y.,
Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol.
2011;
7
(1)
:
58-63
.
View Article PubMed Google Scholar -
Park
J.,
Chen
Y.,
Tishkoff
D.X.,
Peng
C.,
Tan
M.,
Dai
L.,
SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell.
2013;
50
(6)
:
919-30
.
View Article PubMed Google Scholar -
Rardin
M.J.,
He
W.,
Nishida
Y.,
Newman
J.C.,
Carrico
C.,
Danielson
S.R.,
SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab.
2013;
18
(6)
:
920-33
.
View Article PubMed Google Scholar -
Du
J.,
Zhou
Y.,
Su
X.,
Yu
J.J.,
Khan
S.,
Jiang
H.,
Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science.
2011;
334
(6057)
:
806-9
.
View Article PubMed Google Scholar -
Parikh
S.,
Cohen
B.H.,
Gupta
A.,
Lachhwani
D.K.,
Wyllie
E.,
Kotagal
P.,
Metabolic testing in the pediatric epilepsy unit. Pediatr Neurol.
2008;
38
(3)
:
191-5
.
View Article PubMed Google Scholar -
Stacpoole
P.W.,
Lactic acidosis and other mitochondrial disorders. Metabolism.
1997;
46
(3)
:
306-21
.
View Article PubMed Google Scholar -
Maj
M.C.,
MacKay
N.,
Levandovskiy
V.,
Addis
J.,
Baumgartner
E.R.,
Baumgartner
M.R.,
Pyruvate dehydrogenase phosphatase deficiency: identification of the first mutation in two brothers and restoration of activity by protein complementation. J Clin Endocrinol Metab.
2005;
90
(7)
:
4101-7
.
View Article PubMed Google Scholar -
Ferriero
R.,
Iannuzzi
C.,
Manco
G.,
Brunetti-Pierri
N.,
Differential inhibition of PDKs by phenylbutyrate and enhancement of pyruvate dehydrogenase complex activity by combination with dichloroacetate. J Inherit Metab Dis.
2015;
38
(5)
:
895-904
.
View Article PubMed Google Scholar -
Ferriero
R.,
Boutron
A.,
Brivet
M.,
Kerr
D.,
Morava
E.,
Rodenburg
R.J.,
Phenylbutyrate increases pyruvate dehydrogenase complex activity in cells harboring a variety of defects. Ann Clin Transl Neurol.
2014;
1
(7)
:
462-70
.
View Article PubMed Google Scholar -
Stacpoole
P.W.,
Barnes
C.L.,
Hurbanis
M.D.,
Cannon
S.L.,
Kerr
D.S.,
Treatment of congenital lactic acidosis with dichloroacetate. Arch Dis Child.
1997;
77
(6)
:
535-41
.
View Article PubMed Google Scholar -
Falk
R.E.,
Cederbaum
S.D.,
Blass
J.P.,
Gibson
G.E.,
Kark
R.A.,
Carrel
R.E.,
Ketonic diet in the management of pyruvate dehydrogenase deficiency. Pediatrics.
1976;
58
(5)
:
713-21
.
PubMed Google Scholar -
Bough
K.J.,
Rho
J.M.,
Anticonvulsant mechanisms of the ketogenic diet. Epilepsia.
2007;
48
(1)
:
43-58
.
View Article PubMed Google Scholar -
Schwartzkroin
P.A.,
Mechanisms underlying the anti-epileptic efficacy of the ketogenic diet. Epilepsy Res.
1999;
37
(3)
:
171-80
.
View Article PubMed Google Scholar -
Danial
N.N.,
Hartman
A.L.,
Stafstrom
C.E.,
Thio
L.L.,
How does the ketogenic diet work? Four potential mechanisms. J of chil neu.
2013;
28
(8)
:
1027-33
.
View Article PubMed Google Scholar -
Neal
E.,
Dietary treatment of epilepsy: practical implementation of ketogenic therapyJohn Wiley & Sons 2012.
View Article Google Scholar -
Shimazu
T.,
Hirschey
M.D.,
Hua
L.,
Dittenhafer-Reed
K.E.,
Schwer
B.,
Lombard
D.B.,
SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab.
2010;
12
(6)
:
654-61
.
View Article PubMed Google Scholar -
Hirschey
M.D.,
Shimazu
T.,
Goetzman
E.,
Jing
E.,
Schwer
B.,
Lombard
D.B.,
SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature.
2010;
464
(7285)
:
121-5
.
View Article PubMed Google Scholar -
Schwer
B.,
Bunkenborg
J.,
Verdin
R.O.,
Andersen
J.S.,
Verdin
E.,
Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci USA.
2006;
103
(27)
:
10224-9
.
View Article PubMed Google Scholar -
Hallows
W.C.,
Lee
S.,
Denu
J.M.,
Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci USA.
2006;
103
(27)
:
10230-5
.
View Article PubMed Google Scholar -
Laurent
G.,
German
N.J.,
Saha
A.K.,
de Boer
V.C.,
Davies
M.,
Koves
T.R.,
SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell.
2013;
50
(5)
:
686-98
.
View Article PubMed Google Scholar -
Shulga
N.,
Pastorino
J.G.,
Ethanol sensitizes mitochondria to the permeability transition by inhibiting deacetylation of cyclophilin-D mediated by sirtuin-3. J Cell Sci.
2010;
123
(Pt 23)
:
4117-27
.
View Article PubMed Google Scholar -
Brown
R.M.,
Head
R.A.,
Boubriak
I.I.,
Leonard
J.V.,
Thomas
N.H.,
Brown
G.K.,
Mutations in the gene for the E1β subunit: a novel cause of pyruvate dehydrogenase deficiency. Hum Genet.
2004;
115
(2)
:
123-7
.
View Article PubMed Google Scholar
Comments
Downloads
Article Details
Volume & Issue : Vol 7 No 2 (2020)
Page No.: 3645-3658
Published on: 2020-02-29
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 14978 times
- Download PDF downloaded - 2414 times
- View Article downloaded - 0 times
