

Copyrights: Onesimus Mahdi, Mohamad Taufik Hidayat Baharuldin, Nurul Huda Mohd Nor, Samaila Musa Chiroma, Saravanan Jagadeesan, Mohamad Aris Mohd Moklas, 2019. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Alzheimer's disease (AD) is the most frequent and multifactorial form of dementia, characterised by multiple cognitive impairments and personality changes. Different methods including chemicals have been used to induce AD-like symptoms in rodent in order to screen many therapeutic drugs for a variety of cognitive dysfunctions. Articles from reliable databases such as Google Scholar, Science Direct, PubMed, Scopus, and Ovid were searched and retrieved with the following descriptors: 'Alzheimer's Disease', Cognitive impairments', Neurotoxins that induce AD', Alzheimerogenic chemicals', excitotoxins', Amyloid beta', neurofibrillary tangles. A number of chemicals have been studied to develop an animal model of AD on the basis of their mechanism of action for cognitive dysfunctions. Some of such chemicals are Heavy metals, Scopolamine, Ethanol, Colchicine, Streptozotocin, Lipopolysaccharide, and Okadaic acid among others, with a view to understanding the pathogenesis of this devastating disease. The purpose of this review is to put forward some AD pathophysiology including AD causative theories and also highlight some Alzheimerogenic chemicals for the purpose of enriching our existing knowledge. It is worth mentioning that not all the biochemical, histopathological, cognitive and behavioural abnormalities can be recapitulated. Nonetheless, experimental models of AD produced by chemicals offer insights to unravelling the pathogenesis of the disease.
Introduction
As the aging population is increasing, the rate of age-associated diseases among older adults has become a serious health concern worldwide1. It is well-established that these age-related diseases cause progressive and irreversible loss of neurons and subsequently lead to dementia. One of such age-related disorders is Alzheimer’s disease (AD). AD, a neurodegenerative disease, is one of the most common and multifactorial forms of dementia characterized by multiple cognitive impairments, personality changes, and abnormal behavior 2. The key pathological emblems observed in AD brain tissues are amyloid beta (Aβ) peptide and hyperphosphorylated tau (p-tau) protein, although the exact mechanisms which cause these alterations are yet to be uncovered 3. The formation of Aβ is due to the aggregation of extracellular senile plaques (SP), while neurofibrillary tangles (NFTs) are caused by abnormal deposits of p-tau proteins 3.
Furthermore, mitochondrial dysfunction, synaptic damage, inflammatory responses, defective neurotransmissions, hormonal alterations and abnormalities in the cell cycle are all linked with AD 4,5,6. The remarkable risk factors linked with the progression of AD are old age, as well as multiple genetic and environmental factors. Although the etiology of AD remains unclear, multiple findings have revealed that oxidative stress is an early characteristic of the AD pathological process and also involved in the formation of Aβ and NFTs 7. Thus, several attempts are ongoing in order to establish anti-AD drugs that could target specific pathogenesis, but these have not garnered much success. On the other hand, animal models have been used to trigger pathological changes similar to the human form of AD and to identify the pathogenesis, especially during the pre-symptomatic stage8. There is a dearth of knowledge with respect to chemically-induced AD models. Hence, this review is aimed at exploring some of the chemicals that are used for the induction of AD-like pathologies and behavioral deficits.
History of AD
AD was first reported by Alois Alzheimer in 1906, but it was not until around 70 years after its discovery that much interest was placed on AD research 9. The findings that have emerged have unraveled and demonstrated, importantly, that the effects of AD commence a long time before its symptoms manifest 10. Several lines of evidence have revealed that AD is a prevailing root of dementia and a significant source of death in the world 11,12. Since its discovery, AD has received substantial attention among the types of dementias that are of global health concern, over the years, due to its debilitating nature. To date, the precise etiology of AD remains to be elucidated, as well as questions such as why the disorder and its symptoms advance so quickly in some people (but is delayed in others), and how the disorder could be more effectively managed or treated 10.
CHARACTERISTICS AND SYMPTOMS OF AD
AD is characterized by loss of memory, mood swings, problems with attention and orientation, and difficulties in carrying out daily activities. The two main pathological hallmarks of AD are aggregations of amyloid plaques (extracellular) formed by Aβ and NFTs (intracellular), produced by the hyperphosphorylation of tau protein 3. These alterations are ultimately followed by severe damage and loss of neurons in the brain regions concerned with memory and learning 10. AD is distinguished at the cellular level by mitochondrial dysfunction, oxidative stress 13, metal imbalance, inflammation, and apoptosis, among other hallmarks 14,15.
Furthermore, several symptoms are observed in individuals living with AD which changes over a period of time. Of the symptoms reported, loss of memory in recalling recent conversations are some of the early clinical manifestations; as well, depression and apathy have been identified in AD.
Additionally, other symptoms that appear later in life as the disease progresses include amnesia16, disorientation, impaired communication, confusion, poor judgement, difficulty in swallowing, speaking, and walking10. These important changes seen in AD patients reflect the severity of neuronal loss in different regions of the brain. There are three stages in AD: early, moderate and severe- through which the symptoms of the disease progress and differ from individual to individual. During the early stage of AD, patients can carry out basic things independently with little assistance for some activities; these activities include driving, walking, and other hobbies that can still be done by the patients10,17. However, during the moderate stage of AD, patients may be unable to carry out routine activities, may become disoriented, and even develop personality disorders and behavioral changes (such as agitation and suspiciousness). Once it reaches the severe stage of AD, the patients become dependent on people in doing daily activities, such as eating, dressing, and bathing, among other activities17.
Prevalence of AD
AD is the primary root cause of dementia; as of 2015, around 46.8 million people worldwide suffered from dementia18. This number is expected to increase exponentially to reach 131.5 million by 2050 if there are no interventions19,18. In terms of prevalence between genders, AD is more of an old age disease, and women have been reported to have a longer life expectancy when compared to men20. Hence, women account for about two-thirds of the elderly population affected by AD. Indeed, AD has a direct economic burden worldwide, with studies indicating that AD management cost 818 billion USD in 2016; in 2018, the expected projected cost rose to 1 trillion USD21,22.
AD CAUSATIVE HYPOTHESES
TAU HYPOTHESIS
The tau hypothesis suggests that hyperphosphorylation of tau protein leads to the conversion of normal tau into the paired helical filament (PHF-tau) and NFTs23,24,25,26,27,28,29. Previously, Aβ had been the focus of AD research. Only recently have researchers begun to shift focus to tau protein due to the fact that a variety of reports have shown tau proteins being among the key elements contained in the NFTs. Tau protein, a member of the microtubule-associated protein, is also a functional monomeric and unfolded cell membrane protein, located within the cytosol of a neuron and very crucial in tubulin stabilization30. In addition, tau is known to control neurite growth and have a role in axonal guidance, thus enhancing the normal function of neurons31. In humans, tau protein is only found in trace levels in non-neuronal cells32. Tau goes through various post-translational changes, especially hyperphosphorylation, a process which acts as a significant factor in influencing the stability of microtubules, thereby leading to tau protein accumulation in AD30. Previous findings have shown that hyperphosphorylation of tau protein occurs via conformational changes, which are followed by the transformation of tau monomer to tau oligomer, leading to the paired helical filament and NFT formation33,34,35,36,37. Recent studies have revealed that NFT itself does not appear to be implicated in causing neurotoxicity leading to the onset of neurodegeneration38, though the hyperphosphorylated tau that disintegrates to form oligomers (the toxic form of tau) is implicated in neuronal damage39. In addition, recent in vivo and in vitro findings in rats have highlighted that tau pathology may have far-reaching effects in distinct brain regions40,41,39. Tau, a microtubule-associated protein, acts as a scaffolding component to assist in stabilizing microtubules by influencing tubulin stability in order to control the normal neuronal function42,43.
The position of the tau gene, microtubule-associated protein tau (MAPT), is essential and located on 100 kb of the long arm of human chromosome 17 at locus 17q21, and has 16 exons44. Moreover, it has been shown that in the brain of humans, the tau proteins encode six isoforms that have different sizes, with their length range being between 352 and 441 amino acids 45. These isoforms have some variations, three repeats (3R) or four repeats (4R) in the C terminal, and also the presence or absence of one (29 amino acids) or two inserts (58 amino acids) in the N-terminal part, which bind to actin proteins and differentiate them. Findings have revealed that the repeat areas (244-268 amino acids) located in the C-terminal are the main domain, which causes the clinging of tau to microtubule46,47. Furthermore, findings have suggested that tau phosphorylation at specific epitopes could adjust the capability of tau to cling to microtubule48. Many post-translational changes, mostly tau hyperphosphorylation, were considered to be involved as essential elements that influence the assembly of microtubule which, in turn, cause tau accumulation in AD49. Thus, tau protein goes through conformational alterations whereas the transformation of tau monomer to tau oligomer causes the accumulation of tau, thereby causing it to pair with a helical filament and produce NFTs (due to hyperphosphorylation of tau). Emerging studies have revealed that NFTs are not associated with causing neurotoxicity. Nonetheless, the intermediary tau oligomer was reported to be a toxic form of tau that is implicated in synaptic destruction in AD50. Tau pathophysiology is accompanied by abnormality of amyloid precursor protein (APP) and is eminently seen in the brain regions implicated in the memory-hippocampus as well as parts of the cerebral cortex51.
AMYLOID CASCADE HYPOTHESIS
Although the exact etiology of AD remains controversial, the amyloid cascade hypothesis has been widely accepted and is the most well-studied of the hypotheses out there23. The presence of amyloid plaques is, inarguably, a key characteristic of the pathology of AD. The amyloid cascade theory proposes that amyloid plaques, made by the accumulation of Aβ peptides which resulted from the proteolytic separation of APP, are crucial in AD pathology52. Studies have shown that the main composition of amyloid plaques in AD are polypeptides (about 4 kDa) that are usually produced in soluble form. Aβ protein has been shown to have a variety of isoforms, mostly ranging from 39 to 43 amino acids52,53. The two isoforms of APP, APP751, and APP770 are made up of 56 amino acids in their ectodomain.
Furthermore, findings have revealed that Aβ1-40 (Aβ40) and Aβ1-42 (Aβ42) are the two major occurring isoforms54. Abnormal processing of APP has been found to lead to the formation of disproportionate insoluble Aβ isoforms. These assemble and establish aggregates that consist of amyloid protein in the form of oligomers and protofibrils. Indeed, previous findings have suggested that this results from the changing of Aβ monomers to the Aβ oligomers before accumulation55. The oligomers eventually cause damage to the neurons56. Thus, high concentrations of indissoluble and likely Aβ42 oligomers are involved in the synaptic elimination during early stage AD 57.
Formation of Aβ
Aβ peptide 52,58 is a derivative of Amyloid β-precursor protein (AβPP), also an intrinsic type I glycoprotein59 which has a broad ectodomain. The position of the chromosome for AβPP is found on 21q21.2. There are 18 exons in the APP gene that exceeds 170 kb, thereby creating 10 isoforms through discrete splicing. These isoforms measure between 563 and 770 amino acids. APP is 695 amino acids in length and is one of the isoforms that has been previously reported to be found in neurons of the central nervous system 60. The specific area that codes Aβ-strings is made up of 16 and 17 exons, of which therein are (40 and 43) amino acid residues. Specifically, this area continues from the ectodomain to the protein’s transmembrane domain53. APP’s central domain possesses strong affinity; hence, it can bind to ions such as copper (Cu) and Zinc (Zn), as well as heparin and collagen, within the extracellular matrix, thereby mediating the interplay of APP with the extracellular matrix. The sphere implicated in the neuritic process is defined by a precise string which is seen following the inclusion of exon 7 product 61. APP isoforms have been known to play vital roles in enhancing coordination between cells and, thus, a few of those roles are hereby outlined62. APP isoform (of 695 amino acid) has been shown to be involved in aiding coordination between cells and enhancing the connection to the extraneuronal matrix, thereby leading to stability. APP within the intracellular domain may be associated with a cytoskeletal system that transports constituents within a particular cell. APP1-671 (βAPPs) and APP1-687 (αAPPs) are the other two APP isoforms produced in segments; they offer a protective role to the neurons and regulate events at the synapse 44.
Processing of APP
Proteolytic series of events taking place around and in the APP transmembrane sphere yield a better expression for the production of toxic Aβ42 protein, and eventually for AD pathogenesis. Three different divisions release the APP ectodomain from the membrane. These divisions have been confirmed to be α-secretase split (which separates Aβ-domain and inhibits Aβ-development- by generating amyloidogenic substance- due to the incapability of developing pathogenic Aβ) 63, β-secretase cleavage (the segment produced from α-secretase, while the β-secretase segments remains connected to the membrane; following further conversion by γ-secretase it becomes weakened), and Aβ-protein (Aβ42) breakage (whose aggregation is thought to cause AD due to the presence of toxic and fibril aggregates) 52,54,58.
Biochemistry of Senile Plaques
Blocq and Marnesco, in 1892, were the first to demonstrate that senile plaques (SP) could be described as densely packed structures called amyloid bodies. The process by which amyloid bodies develop and are implicated in AD development is known as amyloidosis. Fibrillogenic proteins (about 10 nm in diameter; amyloid fibrils that are smooth and straight) and non-fibril components (ApoE and serum Aβ components) are the contents of the amyloid bodies. The SPs, which are also called amyloid plaques, are the product of an extracellular accumulation of Aβ protein60.
CHOLINERGIC HYPOTHESIS
The oldest theory among the AD causative theories is the cholinergic hypothesis 64. This hypothesis states that a decrease in neurotransmitters, known as acetylcholine, in neurons is responsible for AD etiology. The cholinergic hypothesis has been postulated for more than 3 decades now and suggests that abnormal acetylcholine-containing neurons in the basal ganglia are implicated in cognitive decline seen in AD patients65. Essentially, acetylcholine (ACh) is a neurotransmitter utilized by cholinergic neurons and is crucial for physiological processes like attention, learning, memory, stress response, wakefulness, and sleep, as well as sensory information 66,67,68,69,70. Damage to cholinergic neurons was observed as an important pathological alteration which corresponds with cognitive destruction seen in AD.
Consequently, this hypothesis was initially tested using cholinesterase inhibitors which are used for AD treatment. Thus, as a result of the trial, it has been noted that tacrine, one many cholinesterase inhibitors (including donepezil, galantamine, rivastigmine, and memantine), was the earliest anti-AD drug to be used in the clinic71,72. However, due to some identified severe side effects, it has been retrieved from the market since 2012. Even though cholinesterase blockage is a typical palliative treatment with minor gain, presently, it appears to be the only obtainable clinical remedy that offers hope to despairing patients with AD. Despite these facts, the cholinergic hypothesis has not been widely accepted, mainly due to the fact that the medication was proposed to treat deficiency of acetylcholine but was not effective. Nonetheless, about 4 of the 5 approved anti-AD drugs available in the market today were developed based on the cholinergic hypothesis 23.
OXIDATIVE STRESS HYPOTHESIS
Oxidative stress is believed to be critical in AD pathogenesis 73. Notably, the brain is known to consume more energy and exert more functions than any other organs during mitochondrial respiration, which also increases the likelihood of reactive oxygen species (ROS) exposure74. Furthermore, AD is closely linked to molecular oxidative stress, as well as protein nitration, the rise of protein oxidation, lipid peroxidation, and glycoxidation. Moreover, AD is closely linked to the aggregation of Aβ, which has been reported to cause oxidative stress 75,76,77,78,79,80. Hence, anti-oxidant compounds are good candidates to offer protection against oxidative stress and Aβ toxicity, to a certain extent. Although oxidative stress is one aspect of AD, the antioxidant approach has been disputed, too, since it lacks the potential to impede the advancement of AD, and has been suggested to be used as part of combination therapy 81,82.
INFLAMMATION HYPOTHESIS
Recent studies have uncovered that neuroinflammation and aberrant gliosis are also emblems of AD 83. Indeed, the inflammation hypothesis has been validated by genetic and transcriptomic studies 84,85,86,87. Microglia-related pathways were observed as crucial risk factors for AD and its pathogenesis. A wealth of information has indicated that microglia is a crucial factor. For instance, during the initial phase of AD, microglia and its associated proteins, such as the Triggering Receptor Expressed on Myeloid Cells-2 (TREM2), can influence synaptic reduction 88,89. The process of activity-dependent and long-term synaptic plasticity is the typical and intrinsic molecular basis of learning and memory that could be the observable effects on long-term potentiation90. Thus, subsequently, amyloid plaques will be surrounded by aberrant microglia and astrocytes, and produce several proinflammatory cytokines. These series of events are the steps involved in the evolution of AD. However, in the clinic, non-steroidal anti-inflammatory drugs (NSAIDS) have not appeared to be beneficial. This could be largely due to the link between innate immunity and the complexity of AD pathogenesis; thus, the immune responses generated could either be detrimental or useful based on the context 87,91,92.
Nonetheless, there are new findings that indicate that the PD-1 immune crossing point barrier decreases the pathology of AD, thereby enhancing memory in AD mouse models93,94,95, and has become an area for subsequent research. The recent awareness towards uncovering the mechanisms involved regarding microglia disruption in clipping, neurogenesis and plasticity regulation are unlocking areas, allowing for the exploration of better therapeutic interventions and diagnoses of AD96,97. Understanding the abnormal microglial roles and restoring homeostasis could provide a new set of concepts for treating AD. However, due to the intricacy and distinct roles of microglia in health and disease, new biomarkers that reflect the functions of specific microglia are critically needed92,98.
Despite all the above armamentarium, AD still needs to be properly understood in order to address its remote causes and the mechanisms underlying its progression, of which chemicals or drugs have been indicated as some of the causative factors.
MITOCHONDRIAL CASCADE HYPOTHESIS
Swerdlow was the first person to propose the mitochondrial cascade theory in 2004, where he posits that mitochondrial abnormality is the main reason for Aβ accumulation, the formation of NFT, and degeneration of synapses in AD24. Mitochondrial causative theory utilizes many theoretical liberties. It presumes that the physiologic mechanisms underlying AD and the aging brain are similar. It states that based on the fact that mitochondrial dysfunction in AD is systematic, mitochondrial dysfunction is not enough to represent the effects of neuronal degeneration.
Furthermore, the hypothesis of the mitochondrial cascade forecasts that non-Mendelian inheritance is linked to non-autosomal dominant AD23. Lastly, it postulates that AD brain mitochondrial abnormality propels amyloidosis, phosphorylation of tau, and cell cycle re-entry. Mitochondrial dysfunction is detected in many AD tissues25, which include fibroblasts, platelets, mitochondria, and brain. There are three main defective mitochondrial enzymes involved: α-ketoglutarate dehydrogenase complex, pyruvate dehydrogenase, and cytochrome oxidase26. AD brain investigations have revealed a normal concentration of cytochrome oxidase but with an altered structure of the enzymes itself27. Oxidative stress and proteasome abnormality have been hypothesised to enhance mitochondrial dysfunction28. Moreover, studies involving cytoplasmic hybrids (cybrid) have revealed that mitochondrial DNA (mtDNA) is partly responsible for the decreased cytochrome oxidase in AD29.
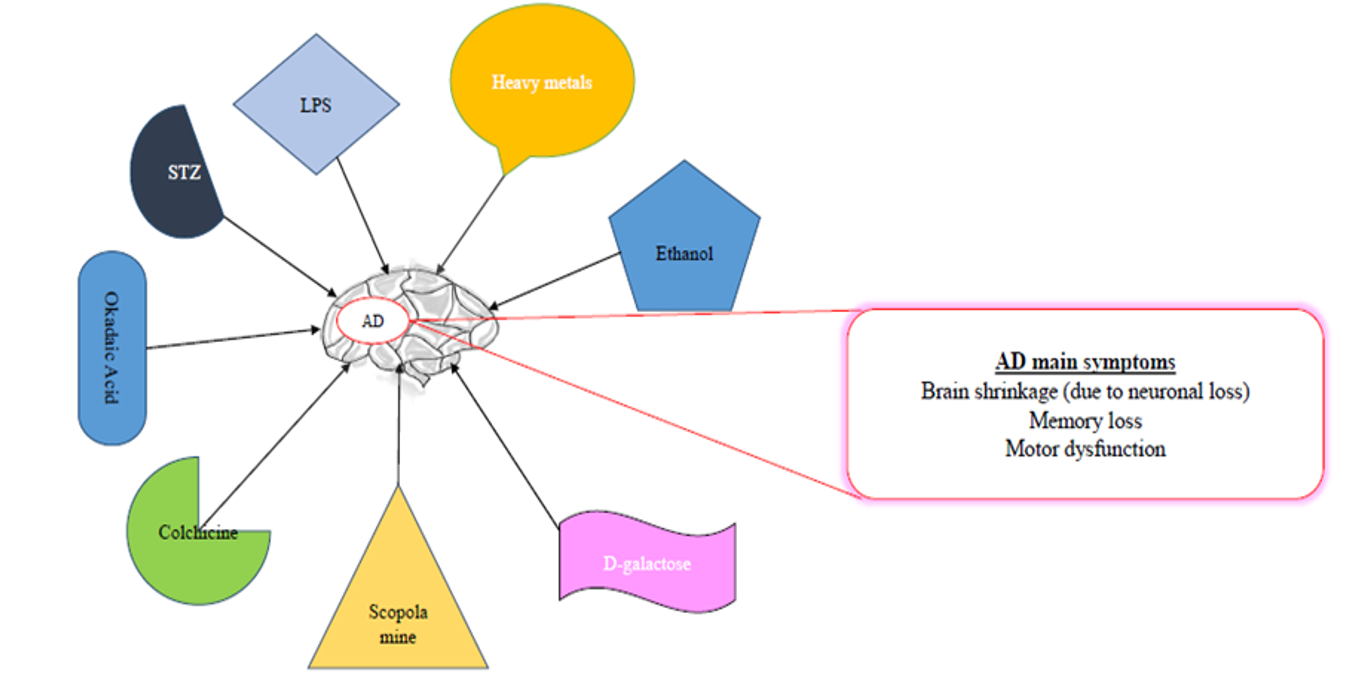
COMMONLY USED CHEMICALS TO INDUCE AD-LIKE SYMPTOMS IN RODENTS
Some of the common chemicals used for modelling AD are scopolamine, streptozotocin, and alcohol, as well as dysregulation of heavy metals, such as aluminum (Al), copper (Cu), zinc (Zn), lead (Pb, and reducing sugar (D-galactose)99, among others (Table 1,Table 2,Table 3).
Aluminum as one of the frequently used heavy metals for induction of cognitive impairment
Environmental heavy metals are agents that have been well-established to impact the development of the brain through neurotoxicity100. Among the commonly used Alzheimerogenic chemicals, heavy metals have been identified to induce high levels of toxicity that is linked with several diseases, including neurodegenerative disorders, following long-term exposure in humans or chronic administration in rodents. A growing body of support has reported the relation between heavy metals and neurodegenerative diseases, including AD and Parkinson’s disease 100. Heavy metals are generally known to damage the nervous system. Of all the heavy metals, the effect of aluminum on biological systems has been well-reported101,45,102. Al is an ubiquitously scattered environmental and industrial toxicant associated with anemia103, osteomalacia, and hepatic and neurological disorders103. The high amount of Al has been identified in the brain of subjects suffering from AD and causes toxicological effects, including encephalopathy, bone disease, and anemia. Very recently, orally administered Al (300 mg/kg body weight) was reported to induce oxidative stress, cholinergic deficit, and accumulation of Aβ & NFTs in the brain of rats101.
Moreover, in the Al-treated group, there are marked histopathological changes and diffuse gliosis accompanied by pericellular edema in the cerebral region, in addition to neuronophagia and loss of neurons. A number of studies have shown that Al neurotoxicity is highly linked with cognitive impairment of AD, via oxidative stress and cholinergic dysfunction104. The findings have indicated an upregulation in the expression and activity of acetylcholinesterase (AChE) and malondialdehyde (MDA), but a significant decrease in expression and activity of glutathione-s-transferase (GST), glutathione peroxides (GPX), and glutathione reductase (GR).
Scopolamine-induced AD-like dementia
A growing body of knowledge has established that scopolamine is an anticholinergic drug mostly employed as an approved chemical in pilot studies to cause memory deficit105. Administration of scopolamine leads to deficits in visual recognition memory, verbal recall, visuospatial praxis, visuospatial recall psychomotor speed, and visuoperceptual processes106. It is one of the most potent and commonly used drugs to prevent motion sickness107. Furthermore, researchers have documented that scopolamine non-selectively occludes the adhesion site of ACh muscarinic receptors in the cerebral cortex and results in the unequal discharge of ACh, which annihilates hippocampal neurons and induces learning and memory impairment in mice in a dose-related manner108,109. However, the main effects of scopolamine likely result from blockage of some receptors (M1 and M5) due to their distinct distribution in the brain110. Previous studies have also suggested that it may lead to memory and learning deficit in a dose-dependent way108,109. In some instances, ordinary doses of scopolamine led to agitation, confusion, hallucination, paranoid behaviors, delusions, and incoherent speech111. Previous studies have indicated there is potential participation of N-methyl-D-aspartate (NMDA) receptor mechanisms of the dorsal hippocampus in memory loss due to scopolamine induction112. Currently, histone deacetylase-2 and DNA methyl transferase-1 are important for synaptic plasticity induced by scopolamine after a decrease in memory113. Hippocampal administration of scopolamine occludes long-term potentiation114 and impedes spatial encoding115.
Furthermore, the injection of scopolamine into the medial septum impedes learning and decreases ACh discharge into the hippocampus116, injection of scopolamine in the CA3 area of the hippocampus induces encoding impairments selectively, but memory retrieval in Hebb-William maze was not affected117. Indeed, scopolamine has been identified to cause spatial learning and memory deficiencies. Consequently, it leads to the excitation of glycogen synthase kinase-3 beta (GSK-3β), inadequate spine maturation, and the arborization of dendrites connected with alterations in CREC, Homer1, and amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPA)118.
Colchicine-induced AD-like symptoms
Advances in chemicals used for modelling AD have suggested that colchicines are potential drugs that induce dementia through dysfunctional cholinergic neurons by either inhibiting cholinergic reversal or eroding cholinergic passage cascades119,120. Colchicine induces hippocampal lesions resulting in cognitive impairments and ChAT reduction, suggesting that it can be utilized as a candidate for modelling AD. Colchicine could cause neurotoxicity and memory decline by inhibiting cholinergic pathways, thereby leading to a reduction in the number of cholinergic neurons and subsequently decreasing cholinergic renewal primarily within the hippocampal area of the brain121. Memory impairment seen induced by colchicine may be a result of a decrease in serotonin, dopamine, and norepinephrine within the caudate nucleus, hippocampus, and entire cerebral cortex122.
Furthermore, colchicine was shown to cause the production of protein carbonyls following lipid peroxidation123. Colchicine has also been found to raise cyclooxygenase-1, and 2 (COX-1 and COX-2) expression levels124 and ROS generation 125. Colchicine raises glutamate/GABA ratio in the cortex126 and triggers excitation of MDA receptors that lead to a sharp rise in the influx of Ca2+, therefore resulting in the triggering of enzymes that depend on Ca2+ (e.g. phospholipases A2, xanthine oxidase, proteases, cyclooxygenases, and protein kinases)127. Intracerebroventricular (ICV) administration of colchicine (7.5 g in 10 L) was found to recapitulate cognitive memory decline in rats128 and similarly in mice119. A significant memory deficit was observed two weeks after cognitive induction impairment using colchicine119. Furthermore, 3 g/mice (ICV injection) of colchicine induces spatial memory impairment129. The main advantage of this model is the fact that it causes some symptoms of sporadic Alzheimer's type of dementia, similar to those found seen in human subjects, like time-variant changes in onset, behavioral, and biological patterns123.
Streptozotocin-induced AD-like cognitive decline
Streptozotocin (STZ), a compound derived from glucosamine nitrosourea and found in the strain of Streptomyces achromogenes, when administered ICV in rodents130, is known to exert a severe and long-lasting effect on the brain’s cytoarchitecture, biochemistry, metabolism, and functions (such as decreased glucose uptake and energy consumption, oxidative tissue stress, cholinergic differential, and cognitive abilities). STZ is another alkylating agent used in cancer treatment. It has also been found to mimic certain properties of nitrosourea, an anti-cancer agent that has been reported to have hypoglycemic effects in addition to being involved in memory damage110. Induction of neuronal damage and hyperphosphorylation of tau is caused by STZ, resulting in the release of ROS and reactive nitrogen species (RNS) 131. Findings revealed that STZ destructs the glycolytic enzyme activity within the brain which eventually results in the lowering of ATP and creatine phosphate concentrations. This destructed energy system and lowered acetyl CoA synthesis can inhibit cholinergic conductance130. Rats induced by STZ have been reported to demonstrate raised activities of AChE in their brains and decreased ACh. In other studies, STZ has been found to activate Aβ peptide-like aggregates by modifying the GSK alpha/beta132.
Furthermore, there is evidence that gene expression associated with the development of glial-derived NF, brain-derived neurotrophic factors (BDNF) and integrin –alpha-M becomes increased by STZ, while expression of genes for NGE-IB and metallothionein 1/2 is down-regulated and eventually results in the modification in apoptosis and cell survival process133. Administration of ICV-STZ resulted in a substantial decrease in memory and learning skills of rats, leading to oxidative stress134. Additionally, ICV administration of STZ (3 mg/kg) in a model of cognitive impairment in rats mimicked the sporadic dementia of AD135. Memory impairment was observed in the rats during the last phase of the experiment using the Morris water maze (MWM) task in order to assess AD-like symptoms. In addition, impaired glucose metabolism, a decrease in cholinergic markers, and oxidative stress were seen in the rats’ brains. Altogether, these long-lasting effects mimicked those seen in AD patients.
| S/N | Species | Chemical | Age/weight/route of administration | Behaviour | Biochemical Assay | Histopathology | Ref |
| 1 | Rats | Al | 150 – 170 g 3 times a week 50 mg/kg 90 days Orally | Short-term & long term cognitive disturbance Slow locomotor activity | ↓ AChE ↓ CAT, GSH | Neuronal loss Vacoulated cytoplasm | 138 |
| 2 | Female Sprague-Dawley Rat | Al. Cu & FlOrally | 170 – 200 g 50, 5 & 20 mg/kg 40 days | Significant learning deficit | ↓ GSH, SOD ↓ GST & GPx↑ proinflammatory TNF-αIL-1β & IL-12 ↑ AChE↑ APP gene expression& Aβ42 level Aβ accumulation,oxidative stress | Degeneration of pyramidal neurons with Pyknotic nuclei Swollen neurons & vacoulation Apoptotic cellsNeuronal degeneration with eisonophilic accumulation | 137 |
| 3 | Rats | Rats AlCl3Orally | 180-200 g300 mg/kg60 days | Memory impairmentSpatial Learning & memory | ↑ AChE activity ↑ proinflammatory cytokines TNF-α,IL-1 & IL-6↑ BDNF mRNA levels↓ CAT, GSH, GST & MDA levels in hippocampus | Neuronal losspyknosis in CA1& CA3 | 2 |
| 4 | Rats | Rats AlCl3Orally | 250-300 g17 mg/kg21 days12—15weeks old | Impaired cognitive functionDecrease in time to reachfood & deteriorated memory | ↑ IL-6 neurotoxicity due to NO & ROS↓ AChE activity↓ ACh↓ BDNF↓ DA↓ TACAβ plaques formation | ↓ in cells in granular& pukinje layersThin irregularreduction in cellcell size of molecular layerSparsely distributed cellsNeurodegeneration in cortex DNA damage | 136 |
| S/N | Species | Chemical | Age/weight/route of administration | Behaviour | Biochemical Assay | Histopathology | Ref |
|---|---|---|---|---|---|---|---|
| 5 | Rats | AlCl3 + D-galOG & IP | 280-300 g10 weeks300 mg/kg60 mg/kg | Memory and LearningdeficitsSpent shorttime in the target quadrant | ↑ AChE activity ↑ Protein expression | Neurodegeneration in hippocampal neuronsAmyloid-like deposit | 103 |
| 6 | Rats | ICV/STZ | 280-300 g0.5 mg/kg(3-5 weeks)1 mg/kg (9-11 weeks) 3 mg/kg | Severe impairment inworking memory | ↓ ChAT mRNA↓ IR expression | Astrogliosisdark neuronsindicating neurodegeneration↓ Astrocytes in DG, CA1 & neuronaldeath | 142 |
| 7 | Rats | ICV/STZ | 350-400 g2 mg/kg3-4 months | Cognitive impairment | ↓ Synaptophysin | Neuronal loss Ventricularenlargementp-tau & Aβ peptide accumulation | 140 |
| 8 | Mice | ICV/STZ Single dose | 20-25 g2 weeks | Learning &memory performance ↓ in mean time spentin a quadrant by MWMDeficit in spatial learningimpairment in novelty inseeking behaviour | ↓ α-secretase activities↓ cerebral Aβ42, β-secretase & COX-2 | Aβ deposits | 139 |
| 9 | Rats | ICV/STZ | 220-25 g5 µl3 months | Deficits in spatial learning & memory in MWM & passive avoidance task | ↑ Oxidative stress↓ GSH ↑ MDA levels | Aβ deposits Shrunken pyramidalcell layer & organisationCellular infiltration& congestionApoptotic cells & neuronal loss | 141 |
| S/N | Species | Chemical | Age/weight/route of administration | Behaviour | Biochemical Assay | Histopathology | Ref |
|---|---|---|---|---|---|---|---|
| 10 | Rats | ICV/STZ | 300-340 g3-4 months30 days | Memory impairmentdecrease in time spentin closed arm 0/OFFDeficits in short-termSpatial memory seenis decrease time spenton new arm Using YMazeShort-term recognition | Neuro inflammation | ↓ Cell propagation↓ in proliferation of marker Ki-67 & immature neurons DCX in SVZ | 154 |
| 11 | Rats | IP/SCM | 150- 250 g20 g/kg | Memory deficit↑ conditional avoidance | ↑ MDA ↑ Lipid peroxidation↓ GSH level↑ AChE activity | Degeneration of neurons with pyknotic &condensed nuclearGliosis | 159 |
| 12 | Rats | IP/SCM | 200-220 g1 mg/kg | Severe memory impairment(ST & LT in hippocampus)Delayed latency in MWM& frontal dependent memoryTask | Occluded cholinergicsignals | Altered cortico-hippocampalneuronsRetraction process in pyramidal cellLayerVacoulation ofsurrounding neutrophilsof pyramidal cells &hyperchromatic &shrunken perikaryo Cork-screw shaped apical dendrites | 169 |
| 13 | Rats | OG/SCM | 150-200 g2.5 mg/kg1 hour | Memory impairment | ↑ AChE activity ↓ GABA &GSH | Neuronal degenerationHippocampal Phalomalacia & Oedema in tissue matrix with demyelination Congestion of blood CapillariesPerivascular Oedema in cortex | 168 |
| 14 | Mice | IP/SCM | 20-25 g1 mg/kg8 weeks old | Impairment of learning & memory Impairment of acquisition of ST & LT | ↓ mRNA expression↔ AChE | - | 167 |
| 15 | Mice | IP/SCM | 17—24 g10 mg/kg7- 12 weeks | Impaired memory↓ ambularv movementDeficits in social recognition | ↑ Cholinergic neurones | - | 166 |
| 16 | Rats/Mice | ICV/CLC | 200-260 g7.5 µg in 2.5 µl aCSF | Memory loss | ↑ TNF-α↑ ROS, COX2Nitrite | - | 165 |
| 17 | Rats | ICV/CLC | 180-200 g15 µ1/5 µl aCSF21 days | Significant memory loss Learning & memory deficits | ↓ GSH, SOD, GST↑ MDA levelsOxidative damage↑ AChE activity | Destruction of neurons | 164 |
| 18 | Rats | ICV/CLC | 180-200 g15 µ1/15 µlaCSF | Cognitive impairment Learning and learning deterioration | ↑ MDA level↔ CAT ↓GSH↓ AChE | Neuronal loss | 163 |
| 19 | Rats | ICV/CLC | 180-200 g15 µ1/rat3 weeks | Memory impairment Impaired acquisitionof spatial navigation task | ↓GSH, Oxidative stress↑ ROS Aβ peptide deposit in hippocampus | Neuronal damage ↓ hippocampal tissuelevels of BDNFAβ deposits in hippocampus | 162 |
| 20 | Rats/Mice | ICV/CLC | 200-250 g 20-30 g 7.5 µl in 2.5 µl 6-8 weeks old | Memory impairment | ↑ TNF-α, IL-113 ↑ ROS & nitrite | Neurodegeneration Plaque formation Reduction in the intensity of Nissl granules | 143 |
| 21 | Rats | IH/OKA | 200-320 g 90 days 100 ng 12 days | Decrease in time finding platformSpatial cognitive | ↓ Glu synthetase ↓ GSHOxidative stress | Gliosis Astrogliosis ↓ GFAP expression | 160 |
| 22 | Rats | ICV/OKA | 300-380 g 100-200 ng | Spatial cognitive deficit Significant ↓ in time to find platform | ↓ GSHOxidative Stress Tau phosphorylation site at 396 | Gliosis p-tau-like formation | 158 |
| 23 | SD Rats | ICV/OKA | 220-250 g 200 ng 13 days | Memory deficitPoor memory performance | ↑ MDA & Nitrite ↓ GSH & Lipid peroxidation | Loss of pyramidal Blabbing of cells in the brain & sponginess Synaptic dysfunction | 157 |
| 24 | Mice | ICV/OKA | 20-22 g 200 ng 2 times 3 days interval | ↓ GPx ↑ MDA mitochondrial cells | Neuronal damage Nuclear pyknosis Reduced density of Nissl bodies Neurofibrillary degeneration | 156 | |
| 25 | Mice | OG/Ethanol | 18-22 g12 mL/gOnce daily1st weekTwice daily after 1st week | Cognitive impairments Short distance covered during spontaneousmovement | ↑ TNF-a &IL-ß ↓ Glu & GABA Neurotransmitter imbalances | Neuronal loss ↓ in microglial cells ↓ hippocampal DG cells | 155 |
| 26 | SD Rats | OG/Ethanol | 257-300 g 5 g/kg Days 2 & 4 | Learning & memorydeficits | ↑ TNF-α inHippocampus ↔ IL-10 BDNF | Shorter microglia Lack of ED-1 positive cells | 170 |
| 27 | Rats | OG/Ethanol | 180-200 g 396-426 g (after long exposure) | Memory impairment | ↑ IL-15 geneNSD alter longterm exposure | Reduction in the microglia shortening & of processes with brush appearance | 153 |
| 28 | SD Rats | SC/Ethanol 4.5 mg/kg | 200-300 g 21 days | ↑ AChE activity | Neuronal death Apoptotic cells Neurodegeneration | 152 | |
| 29 | Rats | SC/Ethanol 4.5 g/kg | 170-220 g 21 days | Cognitive impairment ↓ discrimination index in novels object discrimination | ↓ AChE activity Oxidative stress | - | 151 |
| 30 | SD Rats | ICV/LPS 2 ul/1min | 200-220 g 21 days | Loss of spatial memoryReduction in sniffing timesLess platform-cross number | ↑ IL-IB in hippocampus↑ TNF-e & COX-2↑ NF-Kb, iNOS mRNA | - | 150 |
| 31 | Mice | IH/LPS 40 ug/mouse single admin | 18-22 g 7 days | Learning & memory impairment Treated mice took longertime to find platform usingMWMSpontaneous alteration in Y-Maze | ↓ TNT-α, NO & IL-6Activation of microglia in hippocampus CA1 & DG | Reduction in NeuN stained area of neurons in hippocampus ↓ in number of DCX positive cells | 149 |
| 32 | ICR Mice | IP/LPS 250 ul/kg7 times Daily | - | Memory deficit Spent longer time in a target quadrant | ↓ IL-1β, IL-6 & TNF-α↓ GSH/GSSG↓ COX-2, iNOS↓ MDA content | Brown colouredlabelled (Aβ42) cellsin the hippocampusHigher GFAPIban1-reactive cells | 148 |
| 33 | SD Rats | lP/LPS10 mg/kgSingle dose | 250 g7 —9 days | ↑ TNF-α, IL-1ß &lL-6 | Deposits of AβplaquesP—tau inclusionsin the brain | 147 | |
| 34 | ICR Rats | IP/LPS0.25 mg/kg | 2 months old21 days | Deficits in spatial memoryDecrease in latency | ↑ Aβ342 in cortex& hippocampus↑ β-secretase activity↑ γ-secretase inHippocampus &Cortex↑ iNOS, COX-2 &GFAP | Gliosisin cortex & hippoevidence by thick& short processes | 146 |
| 35 | SD Rats | SC/NAN34—51 mg/kg/day(mini pumps) | 300-320 g31 days | Learning & memory deficitsWeaker cognitive performance | ↓ ChAT & AChEactivity | Pycnotic nervecells Nerve cell loss Corkscrew-like dendritesPositively stained granules by AT8- positive (tau) granule | 145 |
| 36 | Rats | IP/AN312.5 mg/kg/days10 mg/kg/day | 200-250 g5 days9 days | Impairment of learning & memorySpent more time explore targetquadrant | ↓ GSH levels↑ AChE, Nitrite↑ TBARS, MPOCytochrome CNO | Neuronal loss | 161 |
| 37 | SD Rats | SC/NAN3 | 400-425 g1 mg/kg/h7 days | Impairment of learning & memorySpent more time explore targetquadrant | ↓ Cytochrorme C↔ NO | - | 144 |
Ethanol/Alcohol induced AD-like symptoms
Chronic intake of alcohol is associated with many problems including attention deficits, impairment in language and social skills, hyperactivity, motor dysfunction, and learning deficits171. Previous studies have shown that ethanol consumption enhances the generation of ROS and results in a decrease of the antioxidants in the brain172. Another finding has suggested that ethanol could damage hippocampal and cholinergic neurons with a resultant effect on the sensory-motor system as well as disruption of learning and memory173. High ethanol treatment resulted in excessive nitric oxide (NO), which has been found to destruct memory and learning, while higher doses of ethanol disrupted the glutamatergic system and increased GABAergic conveyance in the brain areas that are associated with memory173. Ethanol also elevates the level of adenosine, which may, in turn, lead to memory damage174 and can result in a shorter route by accelerating memory impairment and promoting active, spontaneous motion.
Furthermore, results obtained following the MWM test in mice showed a notable increase in escape latency and total swimming distance, with a sharp drop in cross time. Thus, chronic alcohol administration can significantly affect spatial learning ability and cognition by mice175. However, memory impairment caused by prolonged alcohol intake can be observed from the anti-inflammatory activity and from the control of the equilibrium of Glu and GABA176. The particular molecular mechanisms still need further investigations, but overall, alcohol can significantly disrupt learning and memory.
Memory deficit induced by excitotoxin
Ibotenic acid is a strong neurotoxin that aggravates signs and pathophysiology analogous to AD177,178. It is a useful model to appreciate drug efficiency in evading AD pathology. It has also been found that intrahippocampal administration of ibotenic acid (5 µg/µl PBS) produces memory impairment and results in increased AChE activity as well as elevated MDA levels, thereby inducing neuronal toxicity179. Bilateral ICV injection of ibotenic acid may elicit AD-like pathology and symptoms. Indeed, a particular study found that ibotenic acid decreased the activity of cholinergic neurons in rats180. Many other cholinotoxins and neurotoxins that present AD-like symptoms are kainic acid, quinolinic acid, anti-NGF, NMDA antagonist dizocilpine, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA). Previous studies have reported that intrahippocampal kainic acid (0.4 µg/2µl) administration led to oxidative damage (revealed by the increase of hippocampal lipid peroxidation, nitrite level, and diminished superoxide dismutase (SOD)). Thus, it is clear that kainic acid activates oxidative damage and memory loss in rats 181. Stimulation of glutamate receptors, particularly NMDA receptors, in a diseased condition can lead to disrupted ion homeostasis, decrease in energy, neuronal loss, and cell death (excitotoxicity) by an abnormally high concentration of glutamate 182,183.
Sodium azide-induced AD-like memory impairment
Sodium azide (NaN3) is a white crystalline solid that has been reported to be a mitochondrial toxin implicated in the production of lead azide explosive184. NaN3 administration has been known to induce mitochondrial dysfunction and inhibit cytochrome oxidase, a critical mitochondrial enzyme169. Cytochrome oxidase is critical in the mitochondrial respiratory chain; its interference impedes with mitochondrial complex-IV and decreases ATP levels, which add to metabolic impairment and ROS generation170, ultimately producing some sequelae of AD. Neuronal loss in the CA3 area of the hippocampus is one of the essential features used to confirm the induction of AD using minipumps to deliver NaN3.
However, due to high cost, one-time usability and long treatment time, minipumps are unlikely to be useful tools for screening. A new technique was developed to provide an AD-like dementia model by selective cytochrome C complex inhibition using an intraperitoneal injection of NaN3 in different doses (10-15 mg/kg/day) in rats. The intraperitoneal injection dose-effect-interdependence study employed mitochondrial poison and developed a complex test system to research changes in cognitive functions induced by treatment with NaN3. It has been shown that NaN3 administration impairs learning and memory, and increases AChE levels in the brain169. NaN3 was reported to cause oxidative damage that resulted in neuronal cell death. The progressive loss of neurons and necrosis were observed in the cortical and hippocampal areas of treated rats, thereby further indicating the potency of NaN3. These researchers confirmed that intraperitoneal injection of NaN3 for 15 days resulted in a comparable level of dementia using implanted osmotic minipumps in rats, ultimately suggesting the involvement of NaN3 in inducing neurodegenerative disorder185.
A growing body of knowledge has reported that oxidative stress (Figure 1) is a trigger, as well as one of the critical molecular changes for AD commencement 186. Oxidative stress may be caused by inhibition of mitochondrial function. The mitochondrial enzyme, Cytochrome oxidase (COX), has long been established to be predominantly implicated in aerobic energy metabolism and mitochondrial tasks in AD patients 187. Brains of AD patients have shown some mitochondrial abnormalities, particularly of CoX type. The primary toxic effect of NaN3 has been identified as decreasing the action of CoX in the mitochondrial electron transport chain 185.
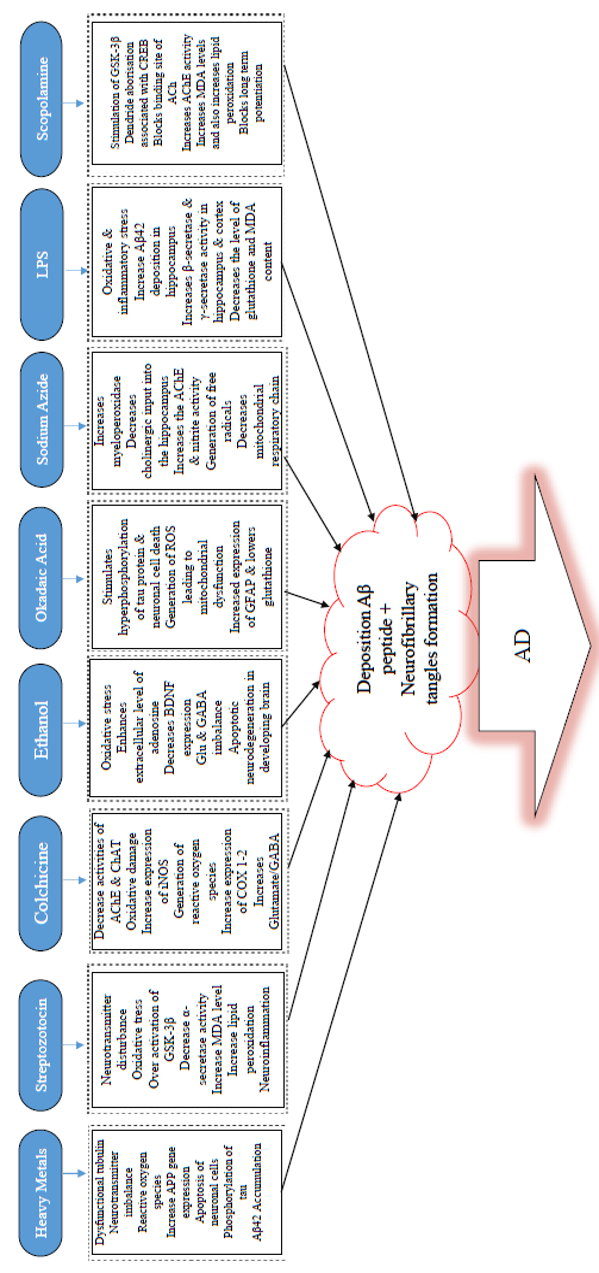
LPS-induced AD-like symptoms
LPS has been known to be used in various experiments, both in vitro and in vivo models of amyloidosis and neuroinflammation 188,189. A variety of neurodegenerative diseases, namely AD 189, Parkinson’s disease190, amyotrophic lateral sclerosis191, and multiple sclerosis192, have been modeled using LPS-induced systemic inflammation (Figure 2). LPS is extracted from the external membrane of gram-negative bacteria. It has been reported as a strong endotoxin with resistivity to degradation by mammalian enzymes, therefore resulting in continual inflammatory stimuli193, which produce proinflammatory cytokines. These proinflammatory cytokines stimulate both neuroimmune and neuroendocrine systems194 that lead to virtually similar feedback formed by behavioral stress195. Inflammation has been reported to be crucial in AD pathogenesis. However, the exact mechanisms through which it participates remains to be elucidated. Inflammatory proteins in the blood, two of which are C reactive protein (CRP) and IL6, have been reported to increase several years before the onset of clinical dementia in different studies12,196. Inflammation is one of the factors that cause AD, although the link between infections and AD etiology has been an issue of debate for more than three decades197. Previous studies have indicated that the expression levels of TNF-α, IL-1β, and IL-6 within the hippocampus are upregulated in comparison with those of control following three days of LPS administration198. Proinflammatory cytokines have already been found as the critical molecules which modulate immune responses; inability to reverse them during chronic inflammation would increase dyshomeostasis198. Moreover, long-standing microglia activation which facilitates inflammatory mediators release, resulting in enhanced oxidative stress and nitrosative stress, is characterized by chronic inflammation 199.
Okadaic acid-induced memory impairment
Okadaic acid (OKA), a major polyether toxin, is a marine microalgae product that causes diarrhetic shellfish toxicity200. IC injection of OKA has been reported to induce memory decline in rats, therefore making it a valuable model agent to study for anti-dementia drug screening201. In terms of mechanism of action, OKA has been found to be a restrictive and modest antagonist of serine/threonine phosphatases 1 and 2A202,203, is associated with short- and long-term memory disruption in rats 204, and triggers tau hyperphosphorylation (Figure 2) and neuronal cell death both in vivo205 and in vitro206. Previous findings have identified that OKA decreases basal synaptic transmission and inhibits the commencement of synaptic plasticity207. Also, OKA has been found to augment Ca2+ in a cultured hippocampal neuronal cells by ionotropic excitatory amino acid receptors, thereby leading to loss of neurons208.
Furthermore, research has demonstrated that OKA triggers the production of ROS in the hippocampus and reduces mitochondrial activity and mitochondrial potency, ultimately resulting in mitochondrial abnormalities in the brain of rats201. Additionally, OKA has been known to inhibit phosphatases and cause hyperphosphorylation of proteins which subsequently leads to neuronal stress and ultimately to neurodegeneration201. Previous findings revealed that OKA-mediated memory decline in rats is linked with exacerbated proinflammatory cytokines, such as TNF-α and IL-1, and with iNOS expression and total nitrite in the hippocampus and cortex 209. Furthermore, infusion (bilateral) of OKA in the hippocampus produced spatial cognitive deficit as a result of raised GFAP expression, decreased GSH, and enhanced protein carbonylation and mitogen-activated protein kinase 38 (p38MAPK)210. Several kinases, including MAPK, GSK-3β and cyclin-dependent kinase 5 (Cdk5), have been identified to be implicated in the phosphorylation of tau protein at different positions seen in AD hyper-phosphorylated tau211,212. Meanwhile, tau dephosphorylation is catalyzed through phosphatase PP2A and other phosphatases (PP1, 2B, and 5)213,214. The precise blocking of PP2A by OKA could result in Alzheimer-like hyperphosphorylation and aggregation of tau in vivo215 and in vitro216,217. Memory loss due to OKA (intra-hippocampal) injection has been documented to be linked with apparent neuropathological changes, such as the formation of Aβ peptide-like structures, helical filament-like phosphorylated tau, and neurodegeneration in the hippocampus210. OKA is an essential means for analyzing218 the cellular mechanisms involved by reversible protein phosphorylation during cell division, signal transduction, and formation of memory217. At present, drugs that act by blocking tau phosphorylation are not available. Thus, this suggests that OKA could be a useful replacement tool towards unravelling therapeutic approaches for AD pathology198.
CONCLUSION
Several chemicals have been established in order to unravel AD etiology and screen many possible therapeutic agents. This applies especially to aging, Aβ, aluminum, and D-galactose models, among others. Nonetheless, it is worthy to note that none of the available models recapitulates the exact pathology of AD, although every model is known to have its benefits and triggers a few of the pathophysiology linked with AD. Heavy metals, scopolamine, alcohol, and LPS are among the chemicals with proven neurotoxicity and have been used to induce AD-like cognitive impairment in rodents. Each chemical may act via specific mechanisms that differ in order to exacerbate AD pathogenesis. Thus, the dosage, as well as the time in which the chemical is administered in the AD models of rodents, may be higher or sometimes lesser to induce severe disease in human subjects. Importantly, other factors associated with the Alzheimerogenic chemicals are the route of administration, nature, duration (especially), as well as age and gender of the animals tested. This understanding is critical in order to design research that involves chemically-induced cognitive impairment.
Abbreviations
AChE: acetyl cholinesterase
ACh: acetylcholine
ChAT: choline acetyltransferase
MDA: Melondialdehyde
SOD: Superoxide dismutase
iNOS: inducible nitric oxide
GSH: glutathione
GPx: glutathione peroxidase
CAT: catalase
DA: dopamine
BDNF: brain derived neurotrophic factors
NO: nitric oxide
ROS: reactive oxygen species
AC: total antioxidant content
OG: oral gavage
ICV: intracerebroventricular
IH: intrahippocampal
SC: subcutaneous
SCM: Scopolamine
STZ: Streptozotocin
OKA: Okadaic acid
CLC: Colchicine
SD: Sprague-Dawley
IP: intraperitoneal
↑: increase
↓: decrease
↔: unchaged
LPS: Lippolyssacharide
TNF-α: Tumor necrosis factor
NFTs: neurofibrillary tangles
COX-2: Cytocrome oxidase
TBARS: Thiobarbituric acid reactive substance
MPO: Myeloperoxidase
GST: Glutathione S-transferase
GFAP: Glial fibrillary acid protein
Conflicts of Interest
Authors disclose none exists.
Authors' Contributions
All authors contributed to the design of the research. OM and SMC searched and summarized the data. SJ, MTBH, NHMN and MAMM reviewed and edited the first draft. All authors reviewed, commented and approved the final draft.
Acknowledgments
The authors would like to acknowledge University Putra Malaysia for sponsoring this project (grant no.GP-IPS 9535400).
References
-
United Nations
Department of Economic,
Social Affairs
Population Division,
World Population Ageing. 2015
.
-
Ravi
S.K.,
Ramesh
B.N.,
Mundugaru
R.,
Vincent
B.,
Multiple pharmacological activities of Caesalpinia crista against aluminium-induced neurodegeneration in rats: relevance for Alzheimer's disease. Environ Toxicol Pharmacol.
2018;
58
:
202-11
.
View Article PubMed Google Scholar -
Tarragon
E.,
Lopez
D.,
Estrada
C.,
Ana
G.C.,
Schenker
E.,
Pifferi
F.,
Octodon degus: a model for the cognitive impairment associated with Alzheimer's disease. CNS Neurosci Ther.
2013;
19
(9)
:
643-8
.
View Article PubMed Google Scholar -
Lam
K.,
Pan
K.,
Linnekamp
J.F.,
Medema
J.P.,
Kandimalla
R.,
DNA methylation based biomarkers in colorectal cancer: A systematic review. Biochim Biophys Acta.
2016;
1866
(1)
:
106-20
.
PubMed Google Scholar -
Wani
W.Y.,
Kandimalla
R.J.,
Sharma
D.R.,
Kaushal
A.,
Ruban
A.,
Sunkaria
A.,
Cell cycle activation in p21 dependent pathway: an alternative mechanism of organophosphate-induced dopaminergic neurodegeneration. Biochim Biophys Acta.
2016;
1863
(7)
:
1858-1866
.
View Article PubMed Google Scholar -
Kandimalla
R.,
Reddy
P.H.,
Multiple faces of dynamin-related protein 1 and its role in Alzheimer's disease pathogenesis. Biochim Biophys Acta.
2016;
1862
(4)
:
814-28
.
View Article PubMed Google Scholar -
Nakagawa
Y.,
Nakamura
S.,
Kaśe
Y.,
Noguchi
T.,
Ishihara
T.,
Colchicine lesions in the rat hippocampus mimic the alterations of several markers in Alzheimer's disease. Brain Res.
1987;
408
(1-2)
:
57-64
.
View Article PubMed Google Scholar -
Li
X.,
Bao
X.,
Wang
R.,
Experimental models of Alzheimer's disease for deciphering the pathogenesis and therapeutic screening (Review). Int J Mol Med.
2016;
37
(2)
:
271-83
.
View Article PubMed Google Scholar -
Alzheimer’s Association Report (2018) Alzheimer’s disease facts and figures Alzheimer’s & Dementia, 14:367-429. 2018
.
-
Villemagne
V.L.,
Burnham
S.,
Bourgeat
P.,
Brown
B.,
Ellis
K.A.,
Salvado
O.,
Australian Imaging Biomarkers
Lifestyle (AIBL) Research Group
Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol.
2013;
12
(4)
:
357-67
.
View Article PubMed Google Scholar -
Katzman
R.,
Editorial: the prevalence and malignancy of Alzheimer disease. A major killer. Arch Neurol.
1976;
33
(4)
:
217-8
.
View Article PubMed Google Scholar -
Zakaria
R.,
Wan Yaacob
W.M.,
Othman
Z.,
Long
I.,
Ahmad
A.H.,
Al-Rahbi
B.,
Lipopolysaccharide-induced memory impairment in rats: a model of Alzheimer's disease. Physiol Res.
2017;
66
(4)
:
553-65
.
PubMed Google Scholar -
Love
S.,
Oxidative stress in brain ischemia. Brain Pathol.
1999;
9
(1)
:
119-31
.
View Article PubMed Google Scholar -
Chin-Chan
M.,
Navarro-Yepes
J.,
Quintanilla-Vega
B.,
Environmental pollutants as risk factors for neurodegenerative disorders: alzheimer and Parkinson diseases. Front Cell Neurosci.
2015;
9
:
124
.
View Article PubMed Google Scholar -
Ramachandran
S.,
Sanjay
S.,
Dhanaraju
M.,
Antiamnesic effect of Piracetam potentiated with Emblica officinalis and Curcuma longa in aluminium induced neurotoxicity of Alzheimer's disease. Int J Adv Res (Indore).
2013;
1
:
185-96
.
-
Kent
B.A.,
Mistlberger
R.E.,
Sleep and hippocampal neurogenesis: implications for Alzheimer's disease. Front Neuroendocrinol.
2017;
45
:
35-52
.
View Article PubMed Google Scholar -
Alistair
B.,
Steve
I.,
CLINICAL REVIEW: alzheimer's disease. BMJ.
2009;
21
(338)
:
467-71
.
-
Alzheimer’s Disease International World Alzheimer Report (2015) The Global Impact of Dementia: An analysis of prevalence, incidence, cost & trends. Pp 1-82. 2015
.
-
Prince
M.,
Wimo
A.,
Guerchet
M.,
Ali
G.C.,
Wu
Y.T.,
Prina
M.,
Alzheimer's Disease International: London; 2015.
Google Scholar -
Luy
M.,
Minagawa
Y.,
Gender gaps life expectancy and proportion of life in poor health. Health Rep.
2014;
25
(12)
:
12-9
.
PubMed Google Scholar -
Wimo
A.,
Guerchet
M.,
Ali
G.C.,
Wu
Y.T.,
Prina
A.M.,
Winblad
B.,
The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimers Dement.
2017;
13
(1)
:
1-7
.
View Article PubMed Google Scholar -
Adlimoghaddam
A.,
Roy
B.,
Albensi
B.C.,
Future Trends and the Economic Burden of Dementia in Manitoba: Comparison with the Rest of Canada and the World. Neuroepidemiology.
2018;
51
(1-2)
:
71-81
.
View Article PubMed Google Scholar -
Mohandas
E.,
Rajmohan
V.,
Raghunath
B.,
Neurobiology of Alzheimer's disease. Indian J Psychiatry.
2009;
51
(1)
:
55-61
.
View Article PubMed Google Scholar -
Swerdlow
R.H.,
Khan
S.M.,
A mitochondrial cascade hypothesis blright for sporadic Alzheimer's disease. Med Hypotheses.
2004;
63
(1)
:
8-20
.
View Article PubMed Google Scholar -
Swerdlow
R.H.,
Kish
S.J.,
Mitochondria in Alzheimer's disease. Int Rev Neurobiol.
2002;
53
:
341-85
.
View Article PubMed Google Scholar -
Gibson
G. E.,
Sheu
K.-F. R.,
Blass
J. P.,
Abnormalities of mitochondrial enzymes in Alzheimer disease. Journal of Neural Transmission.
1998;
105
(8-9)
:
855-870
.
View Article Google Scholar -
Parker
W.D.,
Parks
J.K.,
Cytochrome c oxidase in Alzheimer's disease brain: purification and characterization. Neurology.
1995;
45
(3 Pt 1)
:
482-6
.
View Article PubMed Google Scholar -
Ding
Q.,
Martin
S.,
Dimayuga
E.,
Bruce-Keller
A.J.,
Keller
J.N.,
LMP2 knock-out mice have reduced proteasome activities and increased levels of oxidatively damaged proteins. Antioxid Redox Signal.
2006;
8
(1-2)
:
130-135
.
View Article PubMed Google Scholar -
Swerdlow
R.H.,
Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J Neurosci Res.
2007;
85
(15)
:
3416-28
.
View Article PubMed Google Scholar -
Duan
A.R.,
Jonasson
E.M.,
Alberico
E.O.,
Li
C.,
Scripture
J.P.,
Miller
R.A.,
Interactions between tau and different conformations of tubulin: implications for tau function and mechanism. J Mol Biol.
2017;
429
(9)
:
1424-38
.
View Article PubMed Google Scholar -
Medina
M.,
Avila
J.,
The role of extracellular Tau in the spreading of neurofibrillary pathology. Front Cell Neurosci.
2014;
8
:
113
.
View Article PubMed Google Scholar -
Neselius
S.,
Zetterberg
H.,
Blennow
K.,
Randall
J.,
Wilson
D.,
Marcusson
J.,
Olympic boxing is associated with elevated levels of the neuronal protein tau in plasma. Brain Inj.
2013;
27
(4)
:
425-33
.
View Article PubMed Google Scholar -
Chirita
C.N.,
Congdon
E.E.,
Yin
H.,
Kuret
J.,
Triggers of full-length tau aggregation: a role for partially folded intermediates. Biochemistry.
2005;
44
(15)
:
5862-5872
.
View Article Google Scholar -
Sahara
N.,
Maeda
S.,
Murayama
M.,
Suzuki
T.,
Dohmae
N.,
Yen
S.H.,
Takashima
A.,
Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci.
2007;
25
(10)
:
3020-3029
.
View Article Google Scholar -
Mondragón-Rodríguez
S.,
Basurto-Islas
G.,
Santa-Maria
I.,
Mena
R.,
Binder
L.I.,
Avila
J.,
Cleavage and conformational changes of tau protein follow phosphorylation during Alzheimer’s disease. Int J Exp Pathol.
2008;
89
(2)
:
81-90
.
View Article Google Scholar -
Lasagna-Reeves
C.A.,
Castillo-Carranza
D.L.,
Guerrero-Muñoz
M.J.,
Jackson
G.R.,
Kayed
R.,
Preparation and characterization of neurotoxic tau oligomers. Biochemistry.
2010;
49
(47)
:
10039-10041
.
View Article Google Scholar -
Patterson
K.R.,
Remmers
C.,
Fu
Y.,
Brooker
S.,
Kanaan
N.M.,
Vana
L.,
Ward
S.,
Reyes
J.F.,
Philibert
K.,
Glucksman
M.J.,
Binder
L.I.,
Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease. J Biol Chem.
2011;
286
(26)
:
23063-23076
.
View Article Google Scholar -
Gerson
J.E.,
Castillo-Carranza
D.L.,
Kayed
R.,
Advances in therapeutics for neurodegenerative tauopathies: moving toward the specific targeting of the most toxic tau species. ACS Chem Neurosci.
2014;
5
(9)
:
752-69
.
View Article PubMed Google Scholar -
Guerrero-Muñoz
M.J.,
Gerson
J.,
Castillo-Carranza
D.L.,
Tau Oligomers: The Toxic Player at Synapses in Alzheimer's Disease. Front Cell Neurosci.
2015;
9
:
464
.
View Article PubMed Google Scholar -
Clavaguera
F.,
Bolmont
T.,
Crowther
R.A.,
Abramowski
D.,
Frank
S.,
Probst
A.,
Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol.
2009;
11
(7)
:
909-13
.
View Article PubMed Google Scholar -
Holmes
B.B.,
Diamond
M.I.,
Prion-like properties of tau protein: the importance of extracellular tau as a therapeutic target. J Biol Chem.
2014;
289
(29)
:
19855-19861
.
View Article Google Scholar -
Giacobini
E.,
Gold
G.,
Alzheimer disease therapy moving from amyloid-β to tau. Nat Rev Neurol.
2013;
9
(12)
:
677-86
.
View Article PubMed Google Scholar -
R
Brier M,
B
Gordon,
K
Friedrichsen,
J
McCarthy,
A
Stern,
J
Christensen,
C
Owen,
P
Aldea,
Y
Su,
J
Hassenstab,
NJ
Cairns,
Tau and Aβ imaging, CSF measures, and cognition in Alzheimer's disease. Sci Transl Med.
2016;
11
(8(338))
:
338ra66
.
View Article Google Scholar -
Neve
R.L.,
Harris
P.,
Kosik
K.S.,
Kurnit
D.M.,
Donlon
T.A.,
Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res.
1986;
387
(3)
:
271-80
.
PubMed Google Scholar -
Mujika
J.I.,
Rezabal
E.,
Mercero
J.M.,
Ruipérez
F.,
Costa
D.,
Ugalde
J.M.,
Aluminium in biological environments: a computational approach. Comput Struct Biotechnol J.
2014;
9
(15)
:
e201403002
.
View Article PubMed Google Scholar -
Goedert
M.,
Spillantini
M.G.,
Jakes
R.,
Rutherford
D.,
Crowther
R.A.,
Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron.
1989;
3
(4)
:
519-26
.
View Article PubMed Google Scholar -
Himmler
A.,
Drechsel
D.,
Kirschner
M.W.,
Martin
D.W.,
Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol.
1989;
9
(4)
:
1381-8
.
View Article PubMed Google Scholar -
Callahan
L.M.,
Vaules
W.A.,
Coleman
P.D.,
Progressive reduction of synaptophysin message in single neurons in Alzheimer disease. J Neuropathol Exp Neurol.
2002;
61
(5)
:
384-95
.
View Article PubMed Google Scholar -
Patterson
K.R.,
Remmers
C.,
Fu
Y.,
Brooker
S.,
Kanaan
N.M.,
Vana
L.,
Ward
S.,
Reyes
J.F.,
Philibert
K.,
Glucksman
M.J.,
Binder
L.I.,
Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease. J Biol Chem .
2011;
286
(26)
:
23063-23076
.
View Article Google Scholar -
Fá
M.,
Puzzo
D.,
Piacentini
R.,
Staniszewski
A.,
Zhang
H.,
Baltrons
M.A.,
Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci Rep.
2016;
6
(1)
:
19393
.
View Article Google Scholar -
Hamdane
M.,
Delobel
P.,
Sambo
A.V.,
Smet
C.,
Bégard
S.,
Violleau
A.,
Neurofibrillary degeneration of the Alzheimer-type: an alternate pathway to neuronal apoptosis?. Biochem Pharmacol.
2003;
66
(8)
:
1619-25
.
View Article PubMed Google Scholar -
Hardy
J.,
Selkoe
D.J.,
The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science.
2002;
297
(5580)
:
353-6
.
View Article PubMed Google Scholar -
Skovronsky
D.M.,
Lee
V.M.,
Trojanowski
J.Q.,
Neurodegenerative diseases: new concepts of pathogenesis and their therapeutic implications. Annu Rev Pathol.
2006;
1
(1)
:
151-70
.
View Article PubMed Google Scholar -
Golde
E.T.,
Eckmann
B.C.,
Younkin
S.G.,
ochemical detection of Aβ isoforms: implications for pathogenesis, diagnosis and treatment of Alzheimer’s disease. Biochim Biophys Acta.
2000;
26
(1502(1))
:
172-87
.
View Article Google Scholar -
Kirkitadze
M.D.,
Bitan
G.,
Teplow
D.B.,
Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res.
2002;
69
(5)
:
567-77
.
View Article PubMed Google Scholar -
Kayed
R.,
Head
E.,
Thompson
J.L.,
McIntire
T.M.,
Milton
S.C.,
Cotman
C.W.,
Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science.
2003;
300
(5618)
:
486-9
.
View Article PubMed Google Scholar -
Sanchez-Varo
R.,
Trujillo-Estrada
L.,
Sanchez-Mejias
E.,
Torres
M.,
Baglietto-Vargas
D.,
Moreno-Gonzalez
I.,
Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer's mice hippocampus. Acta Neuropathol.
2012;
123
(1)
:
53-70
.
View Article PubMed Google Scholar -
Suh
Y.H.,
Checler
F.,
Amyloid precursor protein, presenilins, and α-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease. Pharmacol Rev.
2002;
54
(3)
:
469-525
.
View Article PubMed Google Scholar -
Kang
J.,
Lemaire
H.G.,
Unterbeck
A.,
Salbaum
J.M.,
Masters
C.L.,
Grzeschik
K.H.,
The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature.
1987;
325
(6106)
:
733-6
.
View Article PubMed Google Scholar -
Small
D.H.,
McLean
C.A.,
Alzheimer's disease and the amyloid β protein: what is the role of amyloid?. J Neurochem.
1999;
73
(2)
:
443-9
.
View Article PubMed Google Scholar -
Evin
G.,
Weidemann
A.,
Biogenesis and metabolism of Alzheimer's disease Abeta amyloid peptides. Peptides.
2002;
23
(7)
:
1285-97
.
View Article PubMed Google Scholar -
Pasternak
J.J.,
An introduction to human molecular genetics: mechanisms of inherited diseasesJohn Wiley and Sons: Hoboken (New Jersey); 2005.
View Article Google Scholar -
Devi
L.,
Ohno
M.,
Mitochondrial dysfunction and accumulation of the β-secretase-cleaved C-terminal fragment of APP in Alzheimer's disease transgenic mice. Neurobiol Dis.
2012;
45
(1)
:
417-24
.
View Article PubMed Google Scholar -
Francis
P.T.,
Palmer
A.M.,
Snape
M.,
Wilcock
G.K.,
The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry.
1999;
66
(2)
:
137-47
.
View Article PubMed Google Scholar -
Terry
A.V.,
Buccafusco
J.J.,
The cholinergic hypothesis of age and Alzheimer's disease-related cognitive deficits: recent challenges and their implications for novel drug development. J Pharmacol Exp Ther.
2003;
306
(3)
:
821-7
.
View Article PubMed Google Scholar -
Hasselmo
M.E.,
Anderson
B.P.,
Bower
J.M.,
Cholinergic modulation of cortical associative memory function. J Neurophysiol.
1992;
67
(5)
:
1230-46
.
View Article PubMed Google Scholar -
Fine
A.,
Hoyle
C.,
Maclean
C.J.,
Levatte
T.L.,
Baker
H.F.,
Ridley
R.M.,
Learning impairments following injection of a selective cholinergic immunotoxin, ME20.4 IgG-saporin, into the basal nucleus of Meynert in monkeys. Neuroscience.
1997;
81
(2)
:
331-43
.
View Article PubMed Google Scholar -
Sarter
M.,
Bruno
J.P.,
Cognitive functions of cortical acetylcholine: toward a unifying hypothesis. Brain Res Brain Res Rev.
1997;
23
(1-2)
:
28-46
.
View Article PubMed Google Scholar -
Miranda
M.I.,
Bermúdez-Rattoni
F.,
Reversible inactivation of the nucleus basalis magnocellularis induces disruption of cortical acetylcholine release and acquisition, but not retrieval, of aversive memories. Proc Natl Acad Sci USA.
1999;
96
(11)
:
6478-82
.
View Article PubMed Google Scholar -
Haam
J.,
Yakel
J.L.,
Cholinergic modulation of the hippocampal region and memory function. J Neurochem.
2017;
142
:
111-21
.
View Article PubMed Google Scholar -
Brinkman
S.D.,
Gershon
S.,
Measurement of cholinergic drug effects on memory in Alzheimer's disease. Neurobiol Aging.
1983;
4
(2)
:
139-45
.
View Article PubMed Google Scholar -
Summers
W.K.,
Viesselman
J.O.,
Marsh
G.M.,
Candelora
K.,
Use of THA in treatment of Alzheimer-like dementia: pilot study in twelve patients. Biol Psychiatry.
1981;
16
(2)
:
145-53
.
PubMed Google Scholar -
Manoharan
S.,
Guillemin
G.J.,
Abiramasundari
R.S.,
Essa
M.M.,
Akbar
M.,
Akbar
M.D.,
The role of reactive oxygen species in the pathogenesis of Alzheimer's disease, Parkinson's disease, and Huntington's disease: a mini review. Oxid Med Cell Longev.
2016;
2016
:
8590578
.
View Article PubMed Google Scholar -
Chen
R.,
Lai
U.H.,
Zhu
L.,
Singh
A.,
Ahmed
M.,
Forsyth
N.R.,
Reactive oxygen species formation in the brain at different oxygen levels: the role of hypoxia inducible factors. Front Cell Dev Biol.
2018;
6
:
132
.
View Article PubMed Google Scholar -
Butterfield
D.A.,
Griffin
S.,
Munch
G.,
Pasinetti
G.M.,
Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer's disease brain exists. J Alzheimers Dis.
2002;
4
(3)
:
193-201
.
View Article PubMed Google Scholar -
Gibson
G.L.,
Allsop
D.,
Austen
B.M.,
Induction of cellular oxidative stress by the beta-amyloid peptide involved in Alzheimer's disease. Protein Pept Lett.
2004;
11
(3)
:
257-70
.
View Article PubMed Google Scholar -
Mohmmad Abdul
H.,
Sultana
R.,
Keller
J.N.,
St Clair
D.K.,
Markesbery
W.R.,
Butterfield
D.A.,
Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid beta-peptide (1-42), HO and kainic acid: implications for Alzheimer's disease. J Neurochem.
2006;
96
(5)
:
1322-35
.
View Article PubMed Google Scholar -
Butterfield
D.A.,
Reed
T.,
Newman
S.F.,
Sultana
R.,
Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med.
2007;
43
(5)
:
658-77
.
View Article PubMed Google Scholar -
Sultana
R.,
Butterfield
D.A.,
Redox proteomics studies of in vivo amyloid beta-peptide animal models of Alzheimer's disease: insight into the role of oxidative stress. Proteomics Clin Appl.
2008;
2
(5)
:
685-96
.
View Article PubMed Google Scholar -
Cheignon
C.,
Tomas
M.,
Bonnefont-Rousselot
D.,
Faller
P.,
Hureau
C.,
Collin
F.,
Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol.
2018;
14
:
450-64
.
View Article PubMed Google Scholar -
Teixeira
J.,
Silva
T.,
Andrade
P.B.,
Borges
F.,
Alzheimer's disease and antioxidant therapy: how long how far?. Curr Med Chem.
2013;
20
(24)
:
2939-52
.
View Article PubMed Google Scholar -
Persson
T.,
Popescu
B.O.,
Cedazo-Minguez
A.,
Oxidative stress in Alzheimer's disease: why did antioxidant therapy fail?. Oxidative medicine and cellular longevity.
2014;
2014
:
427318
.
View Article Google Scholar -
Salter
M.W.,
Stevens
B.,
Microglia emerge as central players in brain disease. Nat Med.
2017;
23
(9)
:
1018-27
.
View Article PubMed Google Scholar -
Zhang
B.,
Gaiteri
C.,
Bodea
L.G.,
Wang
Z.,
McElwee
J.,
Podtelezhnikov
A.A.,
Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell.
2013;
153
(3)
:
707-20
.
View Article PubMed Google Scholar -
Guerreiro
R.,
Wojtas
A.,
Bras
J.,
Carrasquillo
M.,
Rogaeva
E.,
Majounie
E.,
Alzheimer Genetic Analysis Group
TREM2 variants in Alzheimer's disease. N Engl J Med.
2013;
368
(2)
:
117-27
.
View Article PubMed Google Scholar -
Song
W.,
Hooli
B.,
Mullin
K.,
Jin
S.C.,
Cella
M.,
Ulland
T.K.,
Alzheimer's disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement.
2017;
13
(4)
:
381-7
.
View Article PubMed Google Scholar -
Bolós
M.,
Perea
J.R.,
Avila
J.,
Alzheimer's disease as an inflammatory disease. Biomol Concepts.
2017;
8
(1)
:
37-43
.
View Article PubMed Google Scholar -
Paolicelli
R.C.,
Bolasco
G.,
Pagani
F.,
Maggi
L.,
Scianni
M.,
Panzanelli
P.,
Synaptic pruning by microglia is necessary for normal brain development. Science.
2011;
333
(6048)
:
1456-8
.
View Article PubMed Google Scholar -
Hong
S.,
Dissing-Olesen
L.,
Stevens
B.,
New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol.
2016;
36
:
128-34
.
View Article PubMed Google Scholar -
Bliss
T.V.,
Collingridge
G.L.,
Morris
R.G.,
Synaptic plasticity in health and disease: introduction and overview. Philos Trans R Soc Lond B Biol Sci.
2013;
369
(1633)
:
20130129
.
View Article PubMed Google Scholar -
Chen
J.,
Sun
Z.,
Jin
M.,
Tu
Y.,
Wang
S.,
Yang
X.,
Inhibition of AGEs/RAGE/Rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-κB pathway. J Neuroimmunol.
2017;
305
:
108-14
.
View Article PubMed Google Scholar -
E Hirbec
H.,
Noristani
H.N.,
Perrin
F.E.,
Microglia Responses in Acute and Chronic Neurological Diseases: What Microglia-Specific Transcriptomic Studies Taught (and did Not Teach) Us. Front Aging Neurosci.
2017;
9
:
227
.
View Article PubMed Google Scholar -
Baruch
K.,
Deczkowska
A.,
Rosenzweig
N.,
Tsitsou-Kampeli
A.,
Sharif
A.M.,
Matcovitch-Natan
O.,
PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat Med.
2016;
22
(2)
:
135-7
.
View Article PubMed Google Scholar -
Saresella
M.,
Calabrese
E.,
Marventano
I.,
Piancone
F.,
Gatti
A.,
Calvo
M.G.,
PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis.
2010;
21
(3)
:
927-38
.
View Article PubMed Google Scholar -
S
Marina,
C
Elena,
M
Ivana,
P
Federica,
G
Andrea,
F
Elisabetta,
A potential role for the PD1/PD-L1 pathway in the neuroinflammation of Alzheimer's disease. Neurobiol Aging.
2012;
33
(3)
:
e11-22
.
-
Jevtic
S.,
Sengar
A.S.,
Salter
M.W.,
McLaurin
J.,
The role of the immune system in Alzheimer disease: etiology and treatment. Ageing Res Rev.
2017;
40
:
84-94
.
View Article PubMed Google Scholar -
McGeer
P.L.,
McGeer
E.G.,
Targeting microglia for the treatment of Alzheimer's disease. Expert Opin Ther Targets.
2015;
19
(4)
:
497-506
.
View Article PubMed Google Scholar -
Salter
M.W.,
Stevens
B.,
Microglia emerge as central players in brain disease. Nat Med.
2017;
23
(9)
:
1018-27
.
View Article PubMed Google Scholar -
More
S.V.,
Kumar
H.,
Cho
D.Y.,
Yun
Y.S.,
Choi
D.K.,
Toxin-Induced Experimental Models of Learning and Memory Impairment. Int J Mol Sci.
2016;
17
(9)
:
1447
.
View Article PubMed Google Scholar -
Nallagouni
C.S.,
Reddy
K.P.,
Aluminium and fluoride impacts cortex, hippocampus and dentate gyrus structure in rats: protective effect of resveratrol. Int J Appl Biol Pharm Technol.
2017;
8
:
89-97
.
-
Chiroma
S.M.,
Mohd Moklas
M.A.,
Mat Taib
C.N.,
Baharuldin
M.T.,
Amon
Z.,
d-galactose and aluminium chloride induced rat model with cognitive impairments. Biomed Pharmacother.
2018;
103
:
1602-8
.
View Article PubMed Google Scholar -
Exley
C.,
Human exposure to aluminium. Environ Sci Process Impacts.
2013;
15
(10)
:
1807-16
.
View Article PubMed Google Scholar -
Han
S.,
Lemire
J.,
Appanna
V.P.,
Auger
C.,
Castonguay
Z.,
Appanna
V.D.,
How aluminum, an intracellular ROS generator promotes hepatic and neurological diseases: the metabolic tale. Cell Biol Toxicol.
2013;
29
(2)
:
75-84
.
View Article PubMed Google Scholar -
Calvin
C.,
Systematic review of potential health risks posed by pharmaceutical, occupational and consumer exposures to metallic and nanoscale aluminium, aluminium oxides, aluminium hydroxide and its soluble salts. Crit Rev Toxicol.
2014;
44
(sup4)
:
1-80
.
View Article Google Scholar -
Bubser
M.,
Byun
N.,
Wood
M.R.,
Jones
C.K.,
Muscarinic receptor pharmacology and circuitry for the modulation of cognition. Handb Exp Pharmacol.
2012;
208
(208)
:
121-66
.
View Article PubMed Google Scholar -
Flicker
C.,
Serby
M.,
Ferris
S.H.,
Scopolamine effects on memory, language, visuospatial praxis and psychomotor speed. Psychopharmacology (Berl).
1990;
100
(2)
:
243-50
.
View Article PubMed Google Scholar -
Spinks
A.B.,
Wasiak
J.,
Villanueva
E.V.,
Bernath
V.,
Scopolamine (hyoscine) for preventing and treating motion sickness. Cochrane Database Syst Rev.
2007;
3
(3)
:
002851
.
View Article PubMed Google Scholar -
Riedel
G.,
Kang
S.H.,
Choi
D.Y.,
Platt
B.,
Scopolamine-induced deficits in social memory in mice: reversal by donepezil. Behav Brain Res.
2009;
204
(1)
:
217-25
.
View Article PubMed Google Scholar -
Sreenlvasulu Pattipati
K.A.,
Singh
A.,
Animal Models in Drug Discovery of Alzheimer's Disease: A Mini Review. EC Pharmacology and Toxicology.
2016;
2
(1)
:
60-79
.
-
Klinkenberg
I.,
Blokland
A.,
The validity of scopolamine as a pharmacological model for cognitive impairment: a review of animal behavioral studies. Neurosci Biobehav Rev.
2010;
34
(8)
:
1307-50
.
View Article PubMed Google Scholar -
McEvoy
G.K.,
AHFS drug information. ScopolamineAmerican Society of Health-System Pharmacists: Bethesda (MD); 2005.
Google Scholar -
Khakpai
F.,
Nasehi
M.,
Haeri-Rohani
A.,
Eidi
A.,
Zarrindast
M.R.,
Scopolamine induced memory impairment; possible involvement of NMDA receptor mechanisms of dorsal hippocampus and/or septum. Behav Brain Res.
2012;
231
(1)
:
1-10
.
View Article PubMed Google Scholar -
Singh
P.,
Konar
A.,
Kumar
A.,
Srivas
S.,
Thakur
M.K.,
Hippocampal chromatin-modifying enzymes are pivotal for scopolamine-induced synaptic plasticity gene expression changes and memory impairment. J Neurochem.
2015;
134
(4)
:
642-51
.
View Article PubMed Google Scholar -
Portero-Tresserra
M.,
Del Olmo
N.,
Martí-Nicolovius
M.,
Guillazo-Blanch
G.,
Vale-Martínez
A.,
D-cycloserine prevents relational memory deficits and suppression of long-term potentiation induced by scopolamine in the hippocampus. Eur Neuropsychopharmacol.
2014;
24
(11)
:
1798-807
.
View Article PubMed Google Scholar -
Blokland
A.,
Honig
W.,
Raaijmakers
W.G.,
Effects of intra-hippocampal scopolamine injections in a repeated spatial acquisition task in the rat. Psychopharmacology (Berl).
1992;
109
(3)
:
373-6
.
View Article PubMed Google Scholar -
Elvander
E.,
Schött
P.A.,
Sandin
J.,
Bjelke
B.,
Kehr
J.,
Yoshitake
T.,
Intraseptal muscarinic ligands and galanin: influence on hippocampal acetylcholine and cognition. Neuroscience.
2004;
126
(3)
:
541-57
.
View Article PubMed Google Scholar -
Rogers
J.L.,
Kesner
R.P.,
Cholinergic modulation of the hippocampus during encoding and retrieval. Neurobiol Learn Mem.
2003;
80
(3)
:
332-42
.
View Article PubMed Google Scholar -
Wu
Y.Y.,
Wang
X.,
Tan
L.,
Liu
D.,
Liu
X.H.,
Wang
Q.,
Lithium attenuates scopolamine-induced memory deficits with inhibition of GSK-3β and preservation of postsynaptic components. J Alzheimers Dis.
2013;
37
(3)
:
515-27
.
View Article PubMed Google Scholar -
Kumar
A.,
Seghal
N.,
Padi
S.V.,
Naidu
P.S.,
Differential effects of cyclooxygenase inhibitors on intracerebroventricular colchicine-induced dysfunction and oxidative stress in rats. Eur J Pharmacol.
2006;
551
(1-3)
:
58-66
.
View Article PubMed Google Scholar -
Kumar
A.,
Dogra
S.,
Prakash
A.,
Neuroprotective effects of Centella asiatica against intracerebroventricular colchicine-induced cognitive impairment and oxidative stress. Int J Alzheimers Dis.
2009;
2009
:
972178
.
View Article PubMed Google Scholar -
Evrard
P.A.,
Ragusi
C.,
Boschi
G.,
Verbeeck
R.K.,
Scherrmann
J.M.,
Simultaneous microdialysis in brain and blood of the mouse: extracellular and intracellular brain colchicine disposition. Brain Res.
1998;
786
(1-2)
:
122-7
.
View Article PubMed Google Scholar -
Ganguly
R.,
Guha
D.,
Alteration of brain monoamines & EEG wave pattern in rat model of Alzheimer's disease & protection by Moringa oleifera. Indian J Med Res.
2008;
128
(6)
:
744-51
.
PubMed Google Scholar -
Saini
N.,
Singh
D.,
Sandhir
R.,
Neuroprotective effects of Bacopa monnieri in experimental model of dementia. Neurochem Res.
2012;
37
(9)
:
1928-37
.
View Article PubMed Google Scholar -
Subbaramaiah
K.,
Hart
J.C.,
Norton
L.,
Dannenberg
A.J.,
Microtubule-interfering agents stimulate the transcription of cyclooxygenase-2. Evidence for involvement of ERK1/2 AND p38 mitogen-activated protein kinase pathways. J Biol Chem.
2000;
275
(20)
:
14838-45
.
View Article PubMed Google Scholar -
Kumar
A.,
Dogra
S.,
Prakash
A.,
Protective effect of naringin, a citrus flavonoid, against colchicine-induced cognitive dysfunction and oxidative damage in rats. J Med Food.
2010;
13
(4)
:
976-84
.
View Article PubMed Google Scholar -
Yu
Z.,
Cheng
G.,
Hu
B.,
Mechanism of colchicine impairment on learning and memory, and protective effect of CGP36742 in mice. Brain Res.
1997;
750
(1-2)
:
53-8
.
View Article PubMed Google Scholar -
Sharma
B.,
Singh
N.,
Pitavastatin and 4'-hydroxy-3'-methoxyacetophenone (HMAP) reduce cognitive dysfunction in vascular dementia during experimental diabetes. Curr Neurovasc Res.
2010;
7
(3)
:
180-91
.
View Article PubMed Google Scholar -
Raghavendra
M.,
Maiti
R.,
Kumar
S.,
Acharya
S.,
Role of aqueous extract of Azadirachta indica leaves in an experimental model of Alzheimer's disease in rats. Int J Appl Basic Med Res.
2013;
3
(1)
:
37-47
.
View Article PubMed Google Scholar -
Tota
S.,
Nath
C.,
Najmi
A.K.,
Shukla
R.,
Hanif
K.,
Inhibition of central angiotensin converting enzyme ameliorates scopolamine induced memory impairment in mice: role of cholinergic neurotransmission, cerebral blood flow and brain energy metabolism. Behav Brain Res.
2012;
232
(1)
:
66-76
.
View Article PubMed Google Scholar -
Neha
R.K.,
Sodhi
R.K.,
Jaggi
A.S.,
Singh
N.,
Animal models of dementia and cognitive dysfunction. Life Sci.
2014;
109
(2)
:
73-86
.
View Article PubMed Google Scholar -
Zhou
S.,
Yu
G.,
Chi
L.,
Zhu
J.,
Zhang
W.,
Zhang
Y.,
Neuroprotective effects of edaravone on cognitive deficit, oxidative stress and tau hyperphosphorylation induced by intracerebroventricular streptozotocin in rats. Neurotoxicology.
2013;
38
:
136-45
.
View Article PubMed Google Scholar -
Yang
S.,
Zhou
G.,
Liu
H.,
Zhang
B.,
Li
J.,
Cui
R.,
Protective effects of p38 MAPK inhibitor SB202190 against hippocampal apoptosis and spatial learning and memory deficits in a rat model of vascular dementia. BioMed Res Int.
2013;
2013
:
215798
.
View Article PubMed Google Scholar -
Sodhi
R.K.,
Singh
N.,
Jaggi
A.S.,
Neuroprotective mechanisms of peroxisome proliferator-activated receptor agonists in Alzheimer's disease. Naunyn Schmiedebergs Arch Pharmacol.
2011;
384
(2)
:
115-24
.
View Article PubMed Google Scholar -
Correia
S.C.,
Santos
R.X.,
Santos
M.S.,
Casadesus
G.,
Lamanna
J.C.,
Perry
G.,
Mitochondrial abnormalities in a streptozotocin-induced rat model of sporadic Alzheimer's disease. Curr Alzheimer Res.
2013;
10
(4)
:
406-19
.
View Article PubMed Google Scholar -
Sharma
M.,
Briyal
S.,
Gupta
Y.K.,
Effect of alpha lipoic acid, melatonin and trans resveratrol on intracerebroventricular streptozotocin induced spatial memory deficit in rats. Indian J Physiol Pharmacol.
2005;
49
(4)
:
395-402
.
PubMed Google Scholar -
Borai
I.H.,
Ezz
M.K.,
Rizk
M.Z.,
Aly
H.F.,
El-Sherbiny
M.,
Matloub
A.A.,
Therapeutic impact of grape leaves polyphenols on certain biochemical and neurological markers in AlCl3-induced Alzheimer's disease. Biomed Pharmacother.
2017;
93
:
837-51
.
View Article PubMed Google Scholar -
Hussien
H.M.,
Abd-Elmegied
A.,
Ghareeb
D.A.,
Hafez
H.S.,
Ahmed
H.E.,
El-Moneam
N.A.,
Neuroprotective effect of berberine against environmental heavy metals-induced neurotoxicity and Alzheimer's-like disease in rats. Food Chem Toxicol.
2018;
111
:
432-44
.
View Article PubMed Google Scholar -
Kaddour
T.,
Kharoubi
O.,
Ta\ir
O.A.,
Hellal
N.,
Im'ene
B.,
Aoues
A.,
Aluminium-Induced Acute Neurotoxicity in Rats: treatment with aqueous extract of Arthrophytum (Hammada scoparia). J Acute Dis.
2016;
5
(6)
:
470-82
.
View Article Google Scholar -
AM
El Halawany,
NS
Sayed,
HM
Abdallah,
RS
El Dine,
Protective effects of gingerol on streptozotocin-induced sporadic Alzheimer’s disease: emphasis on inhibition of β-amyloid, COX-2, alpha-, beta-secretases and APH1a. Scientific reports.
2017;
7
(1)
:
2902
.
View Article Google Scholar -
Moreira-Silva
D.,
Carrettiero
D.C.,
Oliveira
A.S.,
Rodrigues
S.,
Dos Santos-Lopes
J.,
Canas
P.M.,
Anandamide Effects in a Streptozotocin-Induced Alzheimer's Disease-Like Sporadic Dementia in Rats. Front Neurosci.
2018;
12
(12)
:
653
.
View Article PubMed Google Scholar -
Samy
Doaa M,
Ismail
Cherine A,
Nassra
Rasha A,
Zeitoun
Teshreen M,
Nomair
Azhar M,
Downstream modulation of extrinsic apoptotic pathway in streptozotocin-induced Alzheimer's dementia in rats: Erythropoietin versus curcumin. European journal of pharmacology.
2016;
770
:
52-60
.
-
Rostami
F.,
Javan
M.,
Moghimi
A.,
Haddad-Mashadrizeh
A.,
Fereidoni
M.,
Streptozotocin-induced hippocampal astrogliosis and insulin signaling malfunction as experimental scales for subclinical sporadic Alzheimer model. Life Sci.
2017;
188
:
172-85
.
View Article PubMed Google Scholar -
Sil
S.,
Ghosh
T.,
Ghosh
R.,
NMDA receptor is involved in neuroinflammation in intracerebroventricular colchicine-injected rats. J Immunotoxicol.
2016;
13
(4)
:
474-89
.
View Article PubMed Google Scholar -
Bennett
M.C.,
Mlady
G.W.,
Kwon
Y.H.,
Rose
G.M.,
Chronic in vivo sodium azide infusion induces selective and stable inhibition of cytochrome c oxidase. J Neurochem.
1996;
66
(6)
:
2606-11
.
View Article PubMed Google Scholar -
Szabados
T.,
Dul
C.,
Majtényi
K.,
Hargitai
J.,
Pénzes
Z.,
Urbanics
R.,
A chronic Alzheimer's model evoked by mitochondrial poison sodium azide for pharmacological investigations. Behav Brain Res.
2004;
154
(1)
:
31-40
.
View Article PubMed Google Scholar -
Dong-Young Choi
Obovatol attenuates LPS-induced memory impairments in mice via inhibition of NF-jB signaling pathway. Neurochem Int.
2013;
60
:
68-77
.
-
Wang
Y.,
Wang
M.,
Xu
M.,
Li
T.,
Fan
K.,
Yan
T.,
Nootkatone, a neuroprotective agent from Alpiniae Oxyphyllae Fructus, improves cognitive impairment in lipopolysaccharide-induced mouse model of Alzheimer's disease. Int Immunopharmacol.
2018;
62
:
77-85
.
View Article PubMed Google Scholar -
Kim
Y. E.,
Hwang
C. J.,
Lee
H. P.,
Kim
C. S.,
Son
D. J.,
Ham
Y. W.,
Inhibitory effect of punicalagin on lipopolysaccharide-induced neuroinflammation, oxidative stress and memory impairment via inhibition of nuclear factor-kappaB. Neuropharmacology.
2017;
117
:
21e32
.
View Article Google Scholar -
Hou
Y.,
Xie
G.,
Miao
F.,
Ding
L.,
Mou
Y.,
Wang
L.,
Pterostilbene attenuates lipopolysaccharide-induced learning and memory impairment possibly via inhibiting microglia activation and protecting neuronal injury in mice. Prog Neuropsychopharmacol Biol Psychiatry.
2014;
54
:
92-102
.
View Article PubMed Google Scholar -
Lee
B.,
Shim
I.,
Lee
H.,
Gypenosides Attenuate Lipopolysaccharide-Induced Neuroinflammation and Memory Impairment in Rats.. Evid Based Complement Alternat Med.
2018;
2018
:
4183670
.
View Article PubMed Google Scholar -
Sahab Uddin
Md.,
Asaduzzaman
Md.,
Neuroprotective Activity of Asparagus racemosus Linn. Against Ethanol- Induced Cognitive Impairment and Oxidative Stress in Rats Brain: Auspicious for Controlling the Risk of Alzheimer's Disease. J Alzheimers Dis Parkinsonism.
2016;
6
(4)
:
1-10
.
-
Lakshmi
K.,
Karishma
S.K.,
Gopi Chand
J.,
Maheswara Reddy
M.,
Narendra Babu
A.,
Bhargav Kumar
N.,
Neuroprotective Activity of Terminalia chebula retz against Ethanol Induced Cognitive Impairment and Oxidative Stress in Rats: Promising for Regimentation the Risk of Alzheimer's Disease. Int J Res Ayurveda Pharm.
2018;
9
(4)
:
90-3
.
-
Cruz
C.,
Meireles
M.,
Silva
S.M.,
Chronic ethanol intake induces partial microglial activation that is not reversed by long-term ethanol withdrawal in the rat hippocampal formation. Neurotoxicology.
2017;
60
:
107-15
.
View Article PubMed Google Scholar -
Bassani
T.B.,
Turnes
J.M.,
Moura
E.L.,
Bonato
J.M.,
Cóppola-Segovia
V.,
Zanata
S.M.,
Effects of curcumin on short-term spatial and recognition memory, adult neurogenesis and neuroinflammation in a streptozotocin-induced rat model of dementia of Alzheimer's type. Behav Brain Res.
2017;
335
:
41-54
.
View Article PubMed Google Scholar -
Cui
S.Q.,
Wang
Q.,
Zheng
Y.,
Xiao
B.,
Sun
H.W.,
Gu
X.L.,
Puerarin protects against damage to spatial learning and memory ability in mice with chronic alcohol poisoning. Braz J Med Biol Res.
2015;
48
(6)
:
515-22
.
View Article PubMed Google Scholar -
Zhang
H.,
Wang
P.,
Hou
H.,
Wen
H.,
Zhou
H.,
Gao
F.,
Histone Modification Is Involved in Okadaic Acid (OA) Induced DNA Damage Response and G2-M Transition Arrest in Maize. PLoS One.
2016;
11
(5)
:
e0155852
.
View Article PubMed Google Scholar -
Kamat
P.K.,
Rai
S.,
Swarnkar
S.,
Shukla
R.,
Nath
C.,
Mechanism of synapse redox stress in Okadaic acid (ICV) induced memory impairment: role of NMDA receptor. Neurochem Int.
2014;
76
:
32-41
.
View Article PubMed Google Scholar -
Broetto
N.,
Hansen
F.,
Brolese
G.,
Batassini
C.,
Lirio
F.,
Galland
F.,
Intracerebroventricular administration of okadaic acid induces hippocampal glucose uptake dysfunction and tau phosphorylation. Brain Res Bull.
2016;
124
:
136-43
.
View Article PubMed Google Scholar -
Palle
S.,
Neerat
P.,
Quercetin nanoparticles attenuates scopolamine induced spatial memory deficits and pathological damages in rats. Bull Fac Pharm Cairo Univ.
2017;
55
(1)
:
101-6
.
View Article Google Scholar -
Costa
A.P.,
Tramontina
A.C.,
Biasibetti
R.,
Batassini
C.,
Lopes
M.W.,
Wartchow
K.M.,
Neuroglial alterations in rats submitted to the okadaic acid-induced model of dementia. Behav Brain Res.
2012;
226
(2)
:
420-7
.
View Article PubMed Google Scholar -
Wong-Riley
M.,
Antuono
P.,
Ho
K.C.,
Egan
R.,
Hevner
R.,
Liebl
W.,
Cytochrome oxidase in Alzheimer's disease: biochemical, histochemical, and immunohistochemical analyses of the visual and other systems. Vision research.
1997;
37
(24)
:
3593-3608
.
-
Mohamed
A.R.,
Soliman
G.Y.,
Ismail
C.A.,
Mannaa
H.F.,
Neuroprotective role of vitamin D3 in colchicine-induced Alzheimer's disease in rats. Alexandria Journal of Medicine..
2015;
51
(2)
:
127-36
.
View Article Google Scholar -
Pitchaimani
V.,
Arumugam
S.,
Thandavarayan
R.A.,
Thiyagarajan
M.K.,
Aiyalu
R.,
Sreedhar
R.,
Nootropic activity of acetaminophen against colchicine induced cognitive impairment in rats. J Clin Biochem Nutr.
2012;
50
(3)
:
241-4
.
View Article PubMed Google Scholar -
Kumar
A.,
Dogra
S.,
Prakash
A.,
Neuroprotective Effects of Centella asiatica against Intracerebroventricular Colchicine-Induced Cognitive Impairment and Oxidative Stress. International Journal of Alzheimer’s Disease.
2009;
2009
:
972178
.
View Article Google Scholar -
Sil
S.,
Ghosh
T.,
Etoricoxib inhibits peripheral inflammation and alters immune responses in intracerebroventricular colchicine injected rats. J Neuroimmunol.
2018;
317
:
15-23
.
View Article PubMed Google Scholar -
Riedel
G.,
Kang
S.H.,
Choi
D.Y.,
Platt
B.,
Scopolamine-induced deficits in social memory in mice: reversal by donepezil. Behav Brain Res.
2009;
204
(1)
:
217-25
.
View Article PubMed Google Scholar -
Lee
G.Y.,
Lee
C.,
G.H. Park,
Jang
J.H.,
Amelioration of Scopolamine-Induced Learning and Memory Impairment by 𝛼-Pinene in C57BL/6 Mice. Evidence-Based Complementary and Alternative Medicine.
2017;
2017
:
4926815
.
-
Zaki
H.F.,
Abd-El-Fattah
M.A.,
Attia
A.S.,
Naringenin protects against scopolamine-induced dementia in rats. Faculty of Pharmacy. Cairo University.
2014;
52
:
15-25
.
-
Imam
A.,
Ajao
M.S.,
Ajibola
M.I.,
Amin
A.,
Abdulmajeed
W.I.,
Lawal
A.Z.,
Black seed oil ameliorated scopolamine-induced memory dysfunction and cortico-hippocampal neural alterations in male Wistar rats. Bull Fac Pharm Cairo Univ.
2016;
54
(1)
:
49-57
.
View Article Google Scholar -
Marshall
S.A.,
Geil
C.R.,
Nixon
K.,
Prior Binge Ethanol Exposure Potentiates the Microglial Response in a Model of Alcohol-Induced Neurodegeneration. Brain Sci.
2016;
6
(2)
:
16
.
View Article PubMed Google Scholar -
Spinetta
M.J.,
Woodlee
M.T.,
Feinberg
L.M.,
Stroud
C.,
Schallert
K.,
Cormack
L.K.,
Alcohol-induced retrograde memory impairment in rats: prevention by caffeine. Psychopharmacology (Berl).
2008;
201
(3)
:
361-71
.
View Article PubMed Google Scholar -
Patil
S.,
Tawari
S.,
Mundhada
D.,
Nadeem
S.,
Protective effect of berberine, an isoquinoline alkaloid ameliorates ethanol-induced oxidative stress and memory dysfunction in rats. Pharmacol Biochem Behav.
2015;
136
:
13-20
.
View Article PubMed Google Scholar -
Mailliard
W.S.,
Diamond
I.,
Recent advances in the neurobiology of alcoholism: the role of adenosine. Pharmacol Ther.
2004;
101
(1)
:
39-46
.
View Article PubMed Google Scholar -
Cui
S.Q.,
Wang
Q.,
Zheng
Y.,
Xiao
B.,
Sun
H.W.,
Gu
X.L.,
Puerarin protects against damage to spatial learning and memory ability in mice with chronic alcohol poisoning. Braz J Med Biol Res.
2015;
48
(6)
:
515-22
.
View Article PubMed Google Scholar -
Zola
S.M.,
Squire
L.R.,
Teng
E.,
Stefanacci
L.,
Buffalo
E.A.,
Clark
R.E.,
Impaired recognition memory in monkeys after damage limited to the hippocampal region. J Neurosci.
2000;
20
(1)
:
451-63
.
View Article PubMed Google Scholar -
Ji
C.,
Li
Q.,
Aisa
H.,
Yang
N.,
Dong
L.,
Gossypium herbaceam extracts attenuate IBO-induced excitotoxicity in rat hippocampus. J. Alzheimer Dis..
2009;
16
:
331-339
.
View Article Google Scholar -
Clark
D.,
Tuor
U.I.,
Thompson
R.,
Institoris
A.,
Kulynych
A.,
Zhang
X.,
Protection against recurrent stroke with resveratrol: endothelial protection. PLoS One.
2012;
7
(10)
:
e47792
.
View Article PubMed Google Scholar -
Karthick
C.,
Periyasamy
S.,
Jayachandran
K.S.,
Anusuyadevi
M.,
Intrahippocampal Administration of Ibotenic Acid Induced Cholinergic Dysfunction via NR2A/NR2B Expression: Implications of Resveratrol against Alzheimer Disease Pathophysiology. Front Mol Neurosci.
2016;
9
(28)
:
28
.
View Article PubMed Google Scholar -
Rattan
A.K.,
Tejwani
G.A.,
The neurotoxic actions of ibotenic acid on cholinergic and opioid peptidergic systems in the central nervous system of the rat. Brain Res.
1992;
571
(2)
:
298-305
.
View Article PubMed Google Scholar -
Kumar
A.,
Prakash
A.,
Pahwa
D.,
Galantamine potentiates the protective effect of rofecoxib and caffeic acid against intrahippocampal Kainic acid-induced cognitive dysfunction in rat. Brain Res Bull.
2011;
85
(3-4)
:
158-68
.
View Article PubMed Google Scholar -
Antzoulatos
E.G.,
Byrne
J.H.,
Learning insights transmitted by glutamate. Trends Neurosci.
2004;
27
(9)
:
555-60
.
View Article PubMed Google Scholar -
Mattson
M.P.,
Pathways towards and away from Alzheimer's disease. Nature.
2004;
430
(7000)
:
631-9
.
View Article PubMed Google Scholar -
Steven
H.,
Anesthetic implications of a Near-Lethal Sodium Azide Exposure Anesth. Analog.
2007;
104
(1)
:
229-30
.
View Article Google Scholar -
Neha
A.,
Bindra
C.S.,
Jain
U.K.,
Investigations on Molecular Mechanism Involved in Neuroprotective Effect of Vitamin D against Sodium Azide Induced Alzheimer's Disease in Rats. World Journal of Pharmaceutical Research.
2016;
5
(6)
:
1154-72
.
-
Gao
C.,
Chang
P.,
Yang
L.,
Wang
Y.,
Zhu
S.,
Shan
H.,
Neuroprotective effects of hydrogen sulfide on sodium azide-induced oxidative stress in PC12 cells. Int J Mol Med.
2018;
41
(1)
:
242-50
.
PubMed Google Scholar -
Cadonic
C.,
Sabbir
M.G.,
Albensi
B.C.,
Mechanisms of mitochondrial dysfunction in Alzheimer's disease. Mol Neurobiol.
2016;
53
(9)
:
6078-90
.
View Article PubMed Google Scholar -
Anaeigoudari
A.,
Soukhtanloo
M.,
Reisi
P.,
Beheshti
F.,
Hosseini
M.,
Inducible nitric oxide inhibitor aminoguanidine, ameliorates deleterious effects of lipopolysaccharide on memory and long term potentiation in rat. Life Sci.
2016;
158
:
22-30
.
View Article PubMed Google Scholar -
Lee
H.J.,
M.K. Park,
Y.R. Seo,
Pathogenic Mechanisms of Heavy Metal Induced-Alzheimer's Disease. Toxicol. Environ. Health..
2018;
10
(1)
:
1-10
.
View Article Google Scholar -
Whitton
P.S.,
Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol.
2007;
150
(8)
:
963-76
.
View Article PubMed Google Scholar -
Zhao
W.,
Xie
W.,
Le
W.,
Beers
D.R.,
He
Y.,
Henkel
J.S.,
Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J Neuropathol Exp Neurol.
2004;
63
(9)
:
964-77
.
View Article PubMed Google Scholar -
Walter
S.,
Doering
A.,
Letiembre
M.,
Liu
Y.,
Hao
W.,
Diem
R.,
The LPS receptor, CD14, in experimental autoimmune encephalomyelitis and multiple sclerosis. Cell Physiol Biochem.
2006;
17
(3-4)
:
167-72
.
View Article PubMed Google Scholar -
Ohanian
S.H.,
Schwab
J.H.,
Persistence of group a streptococcal cell walls related to chronic inflammation of rabbit dermal connective tissue. J Exp Med.
1967;
125
(6)
:
1137-48
.
View Article PubMed Google Scholar -
Maitra
U.,
Deng
H.,
Glaros
T.,
Baker
B.,
Capelluto
D.G.,
Li
Z.,
Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J Immunol.
2012;
189
(2)
:
1014-23
.
View Article PubMed Google Scholar -
Oitzl
M.S.,
van Oers
H.,
Schöbitz
B.,
de Kloet
E.R.,
Interleukin-1 beta, but not interleukin-6, impairs spatial navigation learning. Brain Res.
1993;
613
(1)
:
160-3
.
View Article PubMed Google Scholar -
Sochocka
M.,
Zwolińska
K.,
Leszek
J.,
The infectious etiology of Alzheimer's disease. Curr Neuropharmacol.
2017;
15
(7)
:
996-1009
.
View Article PubMed Google Scholar -
Daulatzai
M.A.,
Fundamental role of pan-inflammation and oxidative-nitrosative pathways in neuropathogenesis of Alzheimer's disease. Am J Neurodegener Dis.
2016;
5
(1)
:
1-28
.
PubMed Google Scholar -
Ghosh
S.,
Lertwattanarak
R.,
Jde
J. Garduño,
Galeana
J.J.,
Li
J.,
Zamarripa
F.,
NMDA receptor is involved in neuroinflammation in intracerebroventricular colchicine-injected rats. Journal of Immunotoxicology.
2015;
13
(4)
:
474-489
.
View Article Google Scholar -
Kamat
P.K.,
Rai
S.,
Nath
C.,
Okadaic acid induced neurotoxicity: an emerging tool to study Alzheimer's disease pathology. Neurotoxicology.
2013;
37
:
163-72
.
View Article PubMed Google Scholar -
Fischer
R.,
Maier
O.,
Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxidative medicine and cellular longevity.
2015;
2015
.
-
Kamat
P.K.,
Tota
S.,
Saxena
G.,
Shukla
R.,
Nath
C.,
Okadaic acid (ICV) induced memory impairment in rats: a suitable experimental model to test anti-dementia activity. Brain Res.
2010;
1309
:
66-74
.
View Article PubMed Google Scholar -
Kamat
P.K.,
Tota
S.,
Shukla
R.,
Ali
S.,
Najmi
A.K.,
Nath
C.,
Mitochondrial dysfunction: a crucial event in okadaic acid (ICV) induced memory impairment and apoptotic cell death in rat brain. Pharmacol Biochem Behav.
2011;
100
(2)
:
311-9
.
View Article PubMed Google Scholar -
Cohen
P.,
Holmes
C.F.,
Tsukitani
Y.,
Okadaic acid: a new probe for the study of cellular regulation. Trends Biochem Sci.
1990;
15
(3)
:
98-102
.
View Article PubMed Google Scholar -
Ishihara
H.,
Martin
B.L.,
Brautigan
D.L.,
Karaki
H.,
Ozaki
H.,
Kato
Y.,
Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun.
1989;
159
(3)
:
871-7
.
View Article PubMed Google Scholar -
Maidana
M.,
Carlis
V.,
Galhardi
F.G.,
Yunes
J.S.,
Geracitano
L.A.,
Monserrat
J.M.,
Effects of microcystins over short- and long-term memory and oxidative stress generation in hippocampus of rats. Chem Biol Interact.
2006;
159
(3)
:
223-34
.
View Article PubMed Google Scholar -
He
J.,
Yamada
K.,
Zou
L.B.,
Nabeshima
T.,
Spatial memory deficit and neurodegeneration induced by the direct injection of okadaic acid into the hippocampus in rats. J Neural Transm (Vienna).
2001;
108
(12)
:
1435-43
.
View Article PubMed Google Scholar -
Cagnoli
C.M.,
Kharlamov
E.,
Atabay
C.,
Uz
T.,
Manev
H.,
Apoptosis induced in neuronal cultures by either the phosphatase inhibitor okadaic acid or the kinase inhibitor staurosporine is attenuated by isoquinolinesulfonamides H-7, H-8, and H-9. J Mol Neurosci.
1996;
7
(1)
:
65-76
.
View Article PubMed Google Scholar -
Koss
D.J.,
Hindley
K.P.,
Riedel
G.,
Platt
B.,
Modulation of hippocampal calcium signalling and plasticity by serine/threonine protein phosphatases. J Neurochem.
2007;
102
(4)
:
1009-23
.
View Article PubMed Google Scholar -
Fernández
M.T.,
Zitko
V.,
Gascón
S.,
Torreblanca
A.,
Novelli
A.,
Neurotoxic effect of okadaic acid, a seafood-related toxin, on cultured cerebellar neurons. Ann N Y Acad Sci.
1993;
679
:
260-9
.
View Article PubMed Google Scholar -
Kamat
P.K.,
Tota
S.,
Rai
S.,
Swarnkar
S.,
Shukla
R.,
Nath
C.,
A study on neuroinflammatory marker in brain areas of okadaic acid (ICV) induced memory impaired rats. Life Sci.
2012;
90
(19-20)
:
713-20
.
View Article PubMed Google Scholar -
Costa
A.P.,
Tramontina
A.C.,
Biasibetti
R.,
Batassini
C.,
Lopes
M.W.,
Wartchow
K.M.,
Neuroglial alterations in rats submitted to the okadaic acid-induced model of dementia. Behav Brain Res.
2012;
226
(2)
:
420-7
.
View Article PubMed Google Scholar -
Liu
M.,
Choi
S.,
Cuny
G.D.,
Ding
K.,
Dobson
B.C.,
Glicksman
M.A.,
Kinetic studies of Cdk5/p25 kinase: phosphorylation of tau and complex inhibition by two prototype inhibitors. Biochemistry.
2008;
47
(32)
:
8367-77
.
View Article PubMed Google Scholar -
Lucas
J.J.,
Hernández
F.,
Gómez-Ramos
P.,
Morán
M.A.,
Hen
R.,
Avila
J.,
Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J.
2001;
20
(1-2)
:
27-39
.
View Article PubMed Google Scholar -
Arendt
T.,
Holzer
M.,
Brückner
M.K.,
Janke
C.,
Gärtner
U.,
The use of okadaic acid in vivo and the induction of molecular changes typical for Alzheimer's disease. Neuroscience.
1998;
85
(4)
:
1337-40
.
View Article PubMed Google Scholar -
Liu
F.,
Grundke-Iqbal
I.,
Iqbal
K.,
Gong
C.X.,
Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci.
2005;
22
(8)
:
1942-50
.
View Article PubMed Google Scholar -
Zhang
Z.,
Simpkins
J.W.,
Okadaic acid induces tau phosphorylation in SH-SY5Y cells in an estrogen-preventable manner. Brain Res.
2010;
1345
:
176-81
.
View Article PubMed Google Scholar -
Alvarez-de-la-Rosa
M.,
Silva
I.,
Nilsen
J.,
Pérez
M.M.,
García-Segura
L.M.,
Avila
J.,
Estradiol prevents neural tau hyperphosphorylation characteristic of Alzheimer's disease. Ann N Y Acad Sci.
2005;
1052
(1)
:
210-24
.
View Article PubMed Google Scholar -
Zhang
Z.,
Simpkins
J.W.,
An okadaic acid-induced model of tauopathy and cognitive deficiency. Brain Res.
2010;
1359
:
233-46
.
View Article PubMed Google Scholar -
Fernández
J.J.,
Candenas
M.L.,
Souto
M.L.,
Trujillo
M.M.,
Norte
M.,
Okadaic acid, useful tool for studying cellular processes. Curr Med Chem.
2002;
9
(2)
:
229-62
.
View Article PubMed Google Scholar
Comments
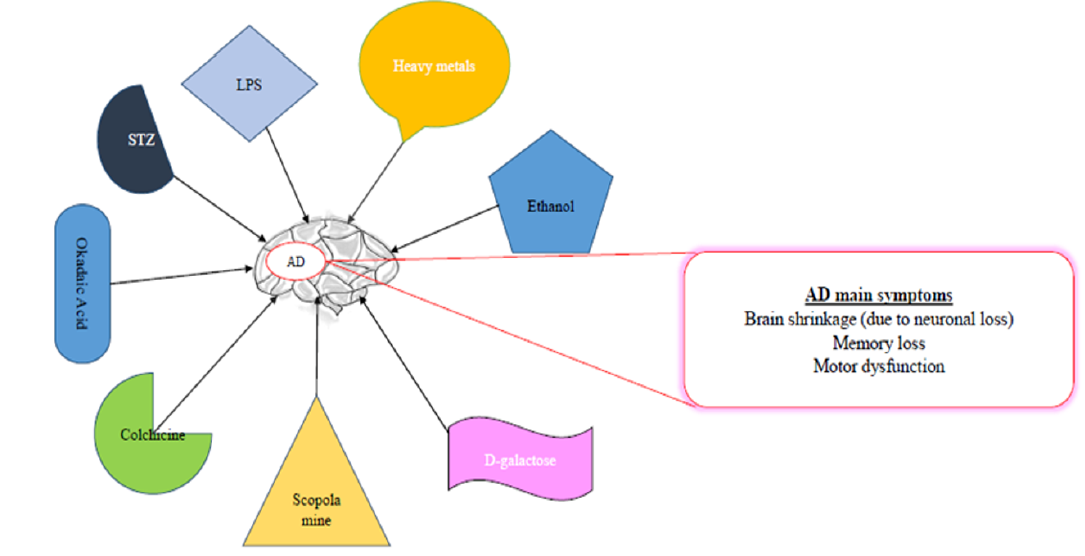
Downloads
Article Details
Volume & Issue : Vol 6 No 11 (2019)
Page No.: 3460-3484
Published on: 2019-11-27
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 32337 times
- Download PDF downloaded - 4889 times
- View Article downloaded - 0 times
