

Molecular Basis of Drug Interactions of Methotrexate, Cyclophosphamide and 5-Fluorouracil as Chemotherapeutic Agents in Cancer
Copyrights: Amit Sarder, Md. Golam Rabbani, A. S. M. Homaun Kabir Chowdhury, Mahbub E Sobhani, 2015. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
At present, chemotherapy is one of the principal methods of treatment of cancer. For many years, chemotherapy is possibly the only way to control cancers that do not respond to either surgery or radiation. To date a good number of chemotherapeutic drugs have been developed which are effective in the treatment of human cancers. But, A few drugs have been known to be safe and promising. The most widely used chemotherapeutic drugs include methotrexate, cyclophosphamide, 5-fluorouracil etc. In this review, the molecular basis of drug interaction of methotrexate, cyclophosphamide and 5-fluorouracil has been studied. Understanding of the molecular basis of drug interaction of the chemotherapeutic agents is fundamental in order to enhance the clinical effectiveness of chemotherapy as well as to increase our knowledge of the cytotoxic effects of chemotherapeutic drugs. Increased understanding of the pharmacokinetics and the mechanism of action also helps to develop biomodulating strategies. The boundary between the efficacy and toxicity of chemotherapeutic drugs is very narrow. So, novel approaches are needed to be formulated in order to enhance the efficacy and to minimize the toxic effects of the chemotherapeutic agents. Though a lot of information is needed to be learned and a lot of task is needed to be accomplished, chemotherapy can be the ultimate and feasible way for controlling cancers if the toxic effects of chemotherapy can be reduced or minimized.
Introduction
Methotrexate
Methotrexate (MTX), 4-amino-N10-methylpteroglutamic acid is a folic acid antagonist first developed for the treatment of malignancies. The IUPAC name of MTX is 4-amino-4-deoxy-10-methyl- pteroyl-glutamic acid and its chemical formula is C20H22N8O5. The molecular weight of MTX is 454.45 Genestier et al., 1998Li, 1960Saxena Ajit. K, 2009. At present, it is most commonly used in the treatment of rheumatoid arthritis (RA). It is now recognized as a very useful disease-modifying anti-rheumatic drug. It is now known that low dose MTX treatment is well- tolerated and produces a quick clinical response in the case of RA patients. It is also very much effective in controlling RA refractory to more conventional treatment Louie and Lillington, 1986Tariq and Tariq,1993. Because of the immunosuppressive, cytostatic and anti-inflammatory effects of MTX, it is used in the systemic treatment of psoriasis. MTX selectively induces apoptosis of activated but not resting lymphocytes, even after short-term exposure to MTX and subsequent activation in drug-free medium. This event provided the first evidence of immunosuppressive activity of low-dose intermittent MTX administration Genestier et al., 1998Kozub and Simaljakova, 2011Saleh Abdulla, 2010. It is also effective in the prevention of acute graft-vs.-host disease (GVHD). But, the combination therapy of MTX with cyclosporine (CSP) has been found to demonstrate superior result than the single MTX therapy Ross et al., 1999Storb et al., 1989von Bueltzingsloewen et al., 1993. Folic acid is an important DNA precursor which undergoes reduction to dihydrofalate and then tetrahydrofolate by the enzyme dihydrofolate reductase (DHFR). MTX and other folate analogues like raltitrexate and pemetrexed act as folate antagonists and inhibit the de novo systhesis of purines. DHFR inhibition by MTX results in reduced folate pool. Reduced folate acts as a cofactor for thymidylate synthase (TS) which catalyzes the synthesis of pyrimidine converting deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP).
Consequently, folate analogues inhibit the synthesis of both purines and pyrimidines, although drugs within this class differ in their relative specificities as inhibitors of DHFR and TS function (Airley, 2009). MTX was the first example of rational drug design and it was mainly developed for the treatment of acute lymphocytic leukaemia. MTX has also been reported to cure choriocarcinoma and trophoblastic cancer Agarwal et al., 2010. MTX can be given in low doses (e.g. 20 mg/m2) in maintenance chemotherapy and also in benign conditions. It has been found that the use of low dose MTX is safe and well tolerated. Because of its safety and efficacy, it is considered as a first line therapy for the treatment of RA. But, MTX can also be given in higher doses (e.g. 1,000 mg/m2 – 33,000 mg/m2) for the treatment of certain cancers. But, high dose MTX therapy can cause significant toxicity like nephrotoxicity, myelosuppression, mucositis, hepatotoxicity etc. In severe cases, it may cause multiorgan failure also Cronstein,2005Rahiem Ahmed YAA, 2013.
Cyclophosphamide
Cyclophosphamide (CP) is one of the most widely used chemotherapeutic agents Juma and Ogada, 1983. It is regarded as a major anticancer drug Rahiem Ahmed YAA, 2013. Though it is a very effective antineoplastic agent against various kinds of tumors, previously it showed virtually no cytotoxic activity against mammalian cell cultures Arthur Camerman, 1977. Basically, CPs are cytotoxic chemotherapy agents similar to mustard gas. It is nitrogen mustard alkylating agent from the oxazophorines group that attaches an alkyl group (CnH2n+1) to Deoxyribonucleotide Acid (DNA). The molecular formula of cyclophosphamide monohydrate is C7H15Q2N2O7P.H2O. The systematic name of cyclophosphamide is 2-[bis(2- Chloroethyl)amino]tetrahydro-2H-1,3,2- oxazaphosphorine 2-oxide monohydrate. CP is mainly used in the treatment of blood cancer, brain cancer, leukemia and some solid tumors Pavan K. V., 2013. It is also used in the treatment of various nonneoplastic diseases like nephritic syndrome, systemic lupus erythematosus and rheumatoid arthritis Morais et al., 1999. As CP is used to treat both neoplastic and non-neoplastic diseases, it is very important to assess the long-term carcinogenic potential of this drug Travis et al., 1995. The side effects of CP are well known in patients receiving this drug for the treatment of lymphomas, leukemias or solid tumours. Bone marrow toxicity with opportunistics infections, haemorrhagic cystitis, temporary infertility, hair loss, vomiting, nausea etc. are seen frequently with the application of CP whereas pneumonitis, liver or cardiac toxicity are very rare (Haubitz, 2007).
5-Fluorouracil
5-Fluorouracil (5-FU) is an example of a rationally designed anticancer drug Malet-Martino and Martino, 2002. This antimetabolite is widely used as a chemotherapeutic drug. Chemically, 5-FU is a dipodic acid and highly polar in nature. 5-FU is a heterocyclic aromatic organic compound with a structure similar to that of the pyrimidine molecules of DNA and RNA. It is basically an analogue of uracil with a fluorine atom at the C-5 position in place of hydrogen. It is one of the most successful chemotherapeutic drugs used for the treatment of solid tumors. It is frequently used as chemotherapeutic agent during the simultaneous chemoradiotrerapy. It is mainly applied in the neoadjuvant and definitive chemoradiotherapy of gastrointestinal malignancies and also in the carcinoma of head and neck. But, fluoropyrimidine therapy is associated with cardiac toxicity. 5-FU induced cardiac toxicity is an underestimated problem in radiooncology. Patients without history of cardiovascular risk factors are often treated as out-patients without cardiac monitoring. Consequently asymptomatic and symptomatic cardiac events may be overlooked. 5-FU is also a drug of choice in oropharyngeal cancer, colorectal cancer, stomach cancer and cervical cancer Lamberti et al., 2012Pendekal and Tegginamat, 2012Steger et al., 2012. It has been found that infusional high dose 5-FU is well-tolerated and effective treatment for gastric cancer. Combination chemotherapy of 5-FU with epirubicin and cisplatin has also resulted in significant response rates and survival benefits for patients with advanced gastric cancer Wang Tso-Fu, 2006. But, clinical studies suggest that the use of 5-FU is not also free from toxicity and women experience more toxicity than men by the 5-FU based chemotherapy. Women not only exhibit a greater variety of toxicity types but also experience them with greater average severity. Common toxicity associated with 5-FU includes stomatitis, leukopenia, alopecia, nausea, vomiting, diarrhea etc.
Molecular basis of drug interaction
Molecular Basis of Drug Interaction of Methotrexate
Tumors are complex tissues of various distinct types of cells. Tumors are more than insular masses of proliferating cancer cells. Tumors are complex tissues of multiple distinct cell types that participate in heterotypic interactions with one another. Also, tumor stroma represents some structural features that are different from the normal tissues. Tumor cell-derived signals activate and recruit hosts cells like monocytes, fibroblasts etc. Thus, the dynamic and reciprocal interactions between tumor cells and the cells of the tumor microenvironment lead to the tumor progression and metastasis formation.
The biology of tumors can no longer be understood by enumerating the traits of the cancer cells but instead must encompass the contributions of the tumor microenvironment to tumorigenesis Hanahan and Weinberg, 2011Ungefroren et al.,2011. MTX can induce apoptosis through both mitochondrial pathway and p53 dependent pathway. These two major mechanisms of action of MTX are discussed below.
Mitochondrial Pathway
Two major classical apoptosis signaling cascades have been known. One major pathway for the initiation of apoptosis is the receptor-mediated pathway or the extrinsic pathway. Another major pathway for the induction of apoptosis is the mitochondrial pathway or the intrinsic pathway Alonso et al., 2005Putcha et al., 2002. The mitochondrial pathway is a complex pathway and acts as central gateway controllers. It has been found that a variety of extracellular and intracellular stress including MTX treatment and oxidative stress can lead to the activation of mitochondrial pathway Chen et al., 2009Lawen, 2003. Oxidative stress can be defined as a state detrimental to the body where oxidative forces exceed the antioxidant systems because of the loss of balance between them. Some atoms and molecules contain unpaired electrons orbiting around the nucleus and these are called free radicals (FR). Free radicals are highly reactive and unstable because the unpaired electrons of free radicals tend to form pairs with other electrons. Oxygen (O2) molecules can undergo 4-electron reduction when it is metabolized in vivo. During this reduction, reactive oxygen metabolites are produced by the excitation of electrons or interaction with transition elements. These reactive oxygen metabolites, also called active oxygen species are highly reactive. Superoxide radical, hydrogen peroxide, alkoxyl radical etc. are examples of active oxygen species. For aerobic organisms, it is very important to have a mechanism of removing these active oxygen species in order to sustain their life. Therefore, various antioxidant defense mechanisms have been developed in the process of evolution. However, if active oxygen species or free radicals are produced excessively, the balance between the formation and removal of active oxygen species are lost. This event results in the oxidative stress Yoshikawa Toshikazu, 2002. Mitochondria are the most important source of reactive oxygen species (ROS) because of the electron transport chain located in the mitochondrial membrane. Again, some cytochrome 450 enzymes can also produce ROS. There are complex interactions between ROS generation, ROS signaling, ROS-induced damage and carcinogenesis. The intrinsic pathway or the mitochondrial pathway can be triggered by many stimuli including ROS. So, the accumulation of ROS can lead to the initiation of apoptosis. As a cytotoxic drug, MTX induces ROS including hydrogen peroxide and superoxide radical and thus induce apoptosis. Hydrogen peroxide accumulated by MTX can cause the release of cytochrome c from the mitochondria into the cytosol. Moreover, it may activate nuclear transcription factors such as activator protein (AP)-1, nuclear factor (NF)-kB and p53 which may lead to the up-regulation of death proteins or production of inhibitors of survival proteins Dayem et al., 2010. AP-1 complex which consists of either Jun homodimers or Fos/Jun heterodimers, bind to a unique palindromic TPA-response element (TRE) with the sequence TGA(C/G)TCA. It is suggested that AP-1 activation is mediated by phosphorylation of Jun proteins. The presence of ROS sensitive kinase cascade, oxidative stress sensitive mitogen-activated protein (MAP) kinase and MAP kinase phosphate is also evident in the activation of AP-1. On the other hand, the activation of NF-kB is thought to be mediated by the phosphorylation of IkB kinase Chung et al., 2002Sen and Packer, 1996. Apoptotic signal from the MTX or the change in the mitochondrial membrane potential(∆ψm) release cytochrome c and other proapoptotic proteins (e.g. adenylate kinase-2 (AK-2), Smac/DIABLO, apoptosis inducing factor (AIF)) from the mitochondrial intermembrane space into the cytosol where cytochrome c binds to a mammalian CED-4 homologue called apoptotic protease activating factor- 1 (Apaf-1). Release of cytochrome c due to the apoptotic signal from the MTX is considered a key step in the apoptotic process. It has been found that the release of cytochrome c occurs by distinct mechanisms that are either Ca2+-dependent or Ca2+-independent. In the Ca2+-dependent mechanism, mitochondrial Ca2+overload promotes the opening of the permeability transition (PT) pore. The opening of this mega channel is linked with the enhanced permeability and loss of mitochondrial membrane potential (∆ψm). This increased inner mitochondrial membrane permeability leads to the swelling of the matrix, rupture of the outer mitochondrial membrane and finally the release of cytochrome c. The release of cytochrome c by the Ca2+- independent mechanism is thought to be governed by different members of the Bcl-2 family of proteins. According to earlier evidences, opening of the mitochondrial permeability pore and loss of mitochondrial membrane potential (∆ψm) is needed for the release of cytochrome c. But, recent data suggest that both events are not needed for the release of apoptotic cytochrome c in all the cases. Binding of cytochrome c to Apaf-1 triggers the formation of the apoptosome which catalyses the activation of caspases. This ~ MDa oligomeric complex contains 7 Apaf-1, 7 cytochrome c, 7 (d)ATP and 7 procaspase-9 molecules. Apaf-1 is basically a 3-domain protein with an N-terminal CARD (caspase recruitment domain) followed by a putative ATPase domain and C-terminal region containing 12 WD40 repeats. Caspase-9, an initiator caspase, is capable of self-processing upon binding with Apaf-1 and it provides a complex for ensuring high local concentration and protein conformation suitable for activation. For the activation of caspase-9, Apaf-1 binds cytochrome c via its WD40 domains. Upon binding to cytochrome c, Apaf-1 becomes competent to recruit caspase-9 in the presence of ADP or ATP. Caspase-9, being activated cleaves and activates pro-caspase-3 Chen et al., 2009Koya et al., 2000Lawen, 2003Ly et al., 2003Ott et al., 2002. It has been found that a compound called PAC-1 activates procaspase-3 to caspase-3. PAC-1 activates procaspase-3 by sequestering inhibitory zinc ions and allows procaspase-3 to auto-activate itself to caspase-3. Thus, PAC-1 induces apoptosis via direct activation of procaspase-3. However, the precise mechanism by which PAC-1 activates procaspase-3 is not yet known. But, it activates procaspase-3 in a dose depended manner. Generally, procaspase-3 levels are high in cancer cells. It suggests that compounds like PAC-1 which stimulates the activation of procaspase-3 to caspase-3 can selectively induce apoptosis. It has been found that the activation of caspase-3 is required for the induction of apoptosis in response to chemotherapeutic agents like MTX. Caspase-3 is the key executioner caspase which catalyzes hydrolysis of a multitude of protein substrate within the cell. Caspase-3 cleaves a variety of cellular substrate including structural proteins and DNA repair enzymes. Caspase-3 mediates proteolysis of several key substrate including the structural proteins gelsolin, focal adhesion kinase, rabaptin-5 and PAK2 and contribute to the drastic morphological change of apoptosis. Caspase-3 also activates an endonuclease caspase-activated DNase (CAD) which causes the fragmentation of DNA by specifically inactivating and cleaving ICAD (DFF45), the inhibitor of CAD. A specific caspase-3 activated DNase, designated as DNA fragmentation factor (DFF40) has been identified. This CAD is revolutionarily conserved crossing rodents and human. CAD/DFF40 exists normally as a nonactive heterodimeric complex in the cell with its natural inhibitor, ICAD (DFF45). Caspase-3 mediated cleavage of ICAD allows the release of CAD/DFF40 from the complex. It is then spontaneously activated. Activated CAD/DFF40 then results in the degradation of genomic DNA into nucleosomal fragments ( Figure 1 ) Cao et al., 2001Janicke et al., 1998McIlroy et al., 2000O’Donovan et al., 2003Peterson et al., 2009.
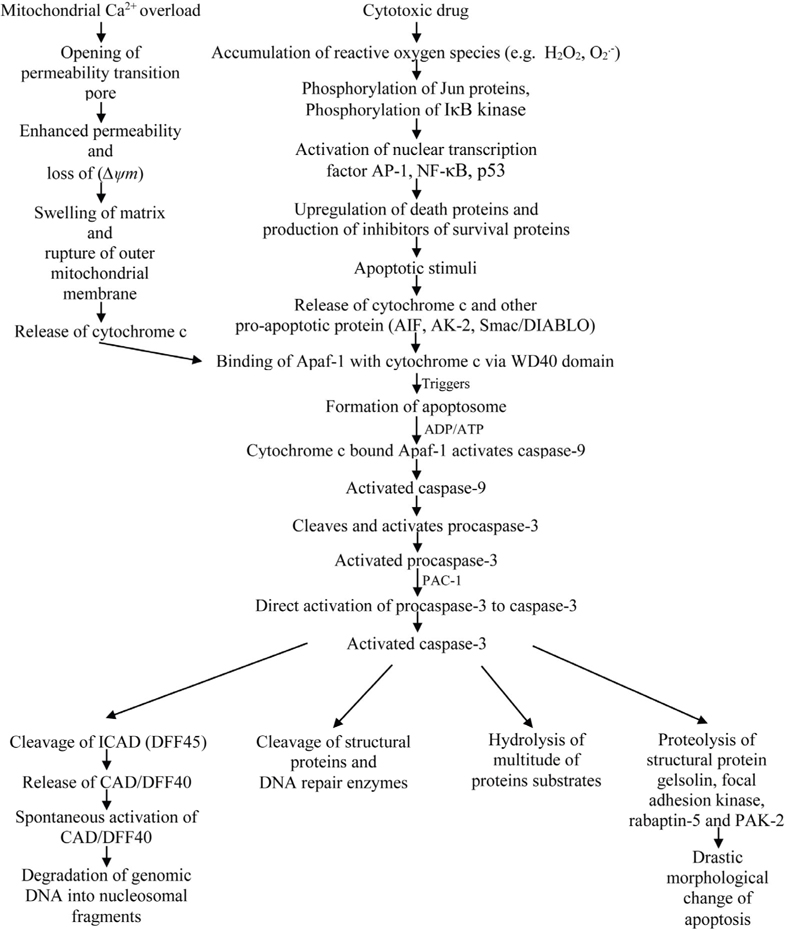
P53 Dependent Pathway
Methotrexate can also induce apoptosis through p53- dependent pathway. The anticancer mechanism of MTX acts through initiation of p53-dependent apoptosis. Methotrexate can induce p53 acetylation at Lys373/382 and phosphorylation at Ser15/Ser392. These events are correlated with the increase in DNA damage and up-regulation of p53 target genes such as p21, DR5, Puma and Noxa. Acetylation of p53 is correlated with its nuclear accumulation and stability. Phosphorylation of p53 also increases its stabilization as well Huang et al., 2011. Cellular stresses like growth factor deprivation, DNA damage or oncogene expression leads to the stabilization and activation of the p53 tumor suppressor protein. This can result in one of the two different outcomes: cell cycle arrest or apoptosis. Cell death induced through p53 pathway is executed by the caspase proteinases. Caspase activation by p53 occurs through the release of apoptogenic factors from mitochondria including cytochrome c. Release of cytochrome c and binding of cytochrome c with the Apaf-1 allows the formation of apoptosome, a high molecular weight complex consisting of Apaf-1 and caspase-9. Caspase-9 is activated following recruitment into the apoptosome. Activated caspase-9 can then cleave and activate the effector caspases like caspase-3 and caspase-7 Schuler and Green, 2001Steele et al., 2008. The activation of these effector caspases leads to the degradation of genomic DNA into nucleosomal fragments, cleavage of structural proteins and DNA repair enzymes, hydrolysis of multitude of protein substrates, drastic morphological change of apoptosis etc. ( Figure 2 ).

Molecular Basis of Drug Interaction of Cyclophosphamide
As an alkylating drug, cyclophosphamide’s therapeutic mechanism of action induces DNA damage. Treatment with CP has also been shown to cause oxidative stress. Tripeptite glutathione (GSH) depletion also induces apoptosis in some cell types in culture. GSH depletion also sensitizes cells to proapoptotic stimuli. CP itself is devoid of alkylating activity and must undergo bio-activation by hepatic cytochrome P450. CP activation is initiated by a cytochrome P450- dependent 4-hydroxylation reaction. CP is metabolically activated in the liver to 4- hydroxycyclophosphamide (4-HC) via oxidation by cytochrome P450 enzymes like CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP3A4 etc. The phenobarbitalinducible rat P450 enzyme 2B1 and its human counterpart 2B6 show high 4-hydroxylase activity compared with other P450 forms. Initially, CP is hydroxylated at C-4 by cytochrome P450. 4-HC then undergoes ring opening to aldo-phosphamide. Aldophosphamide then spontaneously eliminates acrolein and decomposes to the reactive metabolite phosphoramide mustard. Acrolein covalently binds to and there by inactivates several hepatic enzymes including P450. On the other hand, phosphoramide mustard is an active metabolite in terms of both toxicity and anticancer activity. The cytotoxic effects of CP are the result of chemically reactive metabolites that alkylate DNA and protein, producing cross-links. CP, phosphoramide mustard and other metabolites of CP are detoxified by conjugation with GSH Brummaier et al., 2013Cai et al., 1997Chen et al., 2004Clarke and Waxman, 1989Tsai-Turton et al., 2007. The activated CP metabolites, phosphoramide mustard and acrolein are transported into both tumor and healthy cells via the bloodstream. CP and its isomer ifosfamide can induce a caspase-9 dependent apoptotic pathway acting via activated 4-hydroxy metabolites of CP and ifosfamide ( Figure 3 ) Schwartz and Waxman, 2001.
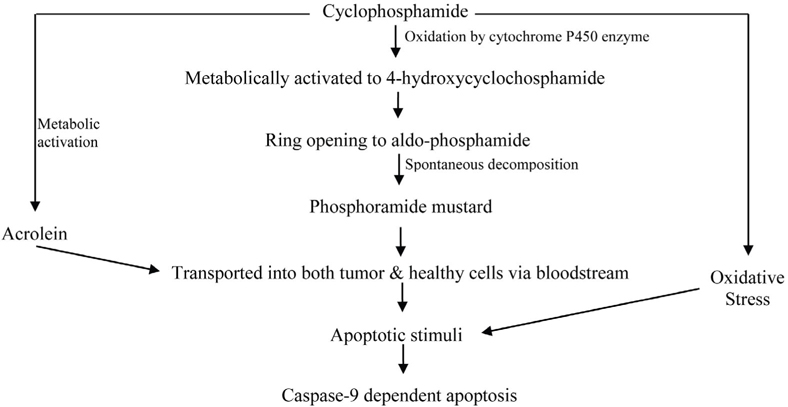
Molecular Basis of Drug Interaction of 5- Fluorouracil
The mechanism of action of 5-FU is associated with the inhibition of TS and incorporation of 5-FU into the RNA and DNA. 5-FU rapidly enters into the cells using the facilitated transport mechanism. 5-FU is then converted into three main active metabolites. The active metabolites are fluorouridine triphosphate (FUTP), fluorodeoxyuridine monophosphate (FdUMP) and fluorodeoxyuridine triphosphate (FdUTP) intracellularly. These three active metabolites disrupt RNA synthesis and the action of TS. The main mechanism of activation of 5-FU is the conversion to fluorouridine monophosphate (FUMP) directly by orotatephosphoribosyltransferase (OPRT) with phosphoribosyl pyrophosphate (PRPP) as the cofactor. This conversion is also caused indirectly by fluorouridine (FUR) in a two-step process involving the action of uridinephosphorylase (UP) and uridine kinase (UK). FUMP is then phosphorylated to fluorouridinediphosphate (FUDP). FUDP can then be further phosphorylated to FUTP which can be incorporated into nascent RNA. FUTP incorporates into the RNA and disrupts the functions of RNA. When FUTP is misincorporated into the RNA, it may interfere the normal nucleic acid metabolism. 5-FU may directly affect the metabolism of RNA. After administration of 5-FU, it can be readily incorporated into the long single rRNa. The incorporation of 5-FU not only inhibit the processing of pre-rRNA into mature rRNA, but also disrupts the post transcriptional modification of tRNAs. It also disrupts the assembly and activity of snRNA/protein complex and thereby inhibits the splicing of pre-mRNa. FUDP can also be converted into the fluorodeoxyuridinediphosphate (FdUDP) by the action of ribonucleotidereductase (RR). FdUDP can then be phosphorylated to yield the active metabolite FdUTP or it can be dephosphorylated to generate the active metabolite FdUMP. 5-FU can also be converted in to the FdUMP in an alternative pathway. 5-FU is converted to the fluorodeoxyuridine (FUDR) by the action of thymidine phosphorylase (TP) which can then be phosphorylated to FdUMP by thymidine kinase (TK). TS is a key enzyme for the de novo synthesis and DNA synthesis. TS catalyses the reductive methylation of dUMP to dTMP. Reduced folate 5, 10- methylenetetrahydrofolate (CH2THF) acts here as the methyl donor. This reaction provides the sole de novo source of thymidylate, which is necessary for the DNA repair and DNA replication. FdUMP, the active metabolite of 5-FU binds to the nucleotide binding site of TS and forms a stable ternary with TS and CH2THF. Thus, it blocks the binding of dUMP and inhibits the synthesis of dTMP. Depletion of dTMP results in the depletion of deoxythymidine triphosphate (dTTP). This event induces the puerturbations in the levels of dATP, dGTP and dCTP also. These results in the deoxynucleotide (dNTP) pool imbalances and increases the levels of dUTP both of which are thought to disrupt DNA synthesis and repair severely leading to the lethal DNA damage. Besides these, TS inhibition results in the depletion of dTTP and an increase in dUTP followed by decreased DNA synthesis and DNA repair. Both dUTP and FdUTP can be misincorporated into DNA. Repair of uracil and 5-FU containing DNA by the nucleotide excision repair enzyme uracil-DNA- glycosylase (UDG) is futile in the presence of high (F)dUTP/dTTP ratios and results in the false nucleotide incorporation. These futile cycles of misincorporation, excision and repair leads to the breakage of DNA strands and cell death. The extent of DNA damage is dependent on the levels of pyrophosphatase dUTPase and UDG. Thymidylate can be salvaged from thymidine through the action of TK ( Figure 4 ) Kaysen et al., 1986Longley et al., 2003Mojardin et al., 2013Roobol et al., 1984Van Triest et al., 2000Zhao and Yu, 2007.
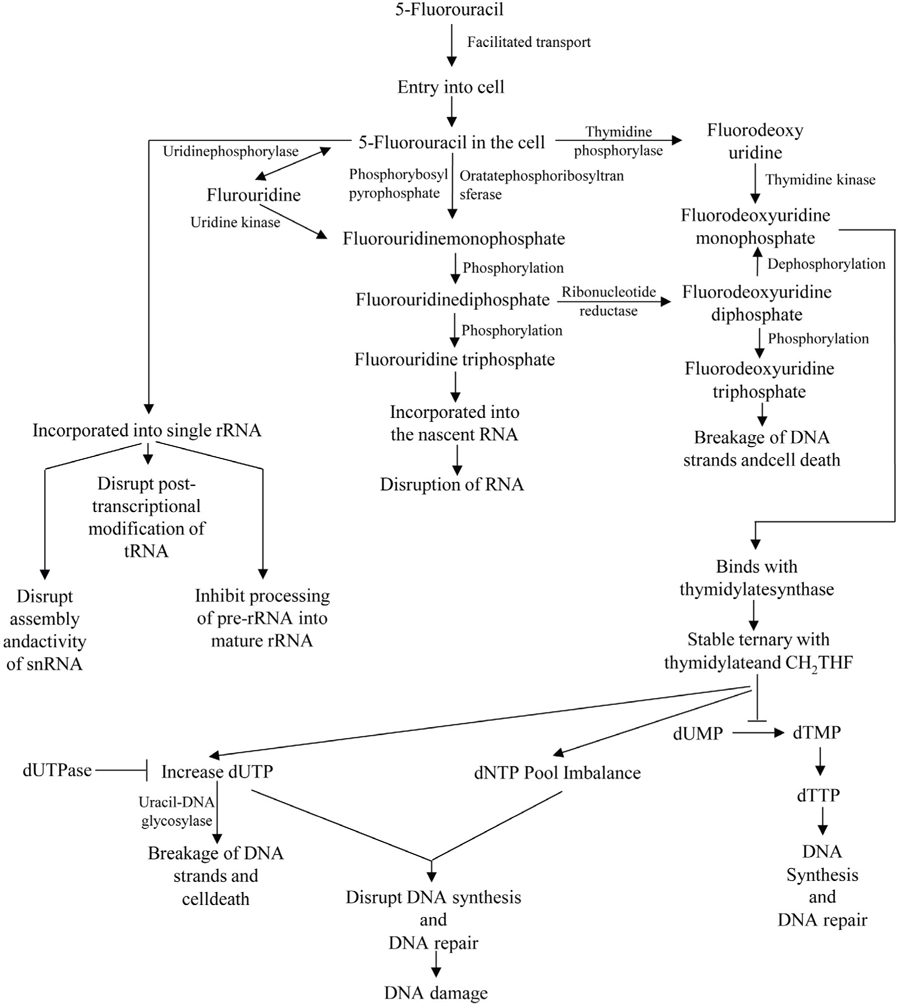
Discussion
Cancer is a life threatening disease and chemotherapy is possibly the only way to control cancers that do not respond to either surgery or radiation. But, little information is available regarding the molecular basis of drug interaction of chemotherapeutic drugs. Understanding of the molecular basis of drug interaction of the commonly used chemotherapeutic drugs is fundamental in order to enhance the clinical effectiveness of these drugs in chemotherapy as well as to increase our knowledge of the cytotoxic effects of chemotherapeutic drugs. Besides, understanding of the molecular basis of drug interaction of the chemotherapeutic drug will lead to the formulation of rationally designed chemotherapeutic agent treatment combinations. Understanding of the mechanisms by which chemotherapeutic drugs induce cell death and the mechanisms by which tumors and cancerous cell become resistant to chemotherapeutic drugs is very crucial in order to overcome drug resistance. Increased understanding of the pharmacokinetics and the mechanism of action also helps to develop bio-modulating strategies. For example, MTX is not only a good chemotherapeutic agent but also a good disease modifying antirheumatic drug. So, the knowledge of molecular basis of drug interaction of MTX will help to use it in combination with other disease modifying anti-rheumatic drug or with newer therapies targeting cells or various inflammatory cytokines. The boundary between the efficacy and the toxicity of the chemotherapeutic drugs is very narrow. So, novel approaches are needed to be formulated in order to enhance the efficacy and to minimize the toxic effects of the chemotherapeutic agents. Construction of structural hybrids of the members of the various gene families or targeting certain active proteins, growth factors, receptors or kinases can be novel approach to treat cancer. As the targets are the cancer specific proteins, the drugs will be comparatively less toxics to the healthy cells and tissues. When chemotherapeutic drugs are administered, the drugs not only interact with the tumor or cancer cells but also it can affect the healthy cells of the body and can cause severe cytotoxic effects. So, the attempts to target drugs to specific sites of the body can be a fruitful way to minimize the cytotoxic effects. Although it is not an easy task to accomplish but the concept of nano-systems in drug targeting can be an option for this. Drug targeting can potentially reduce the toxicity and increase the efficacy of new and the existing drugs. Reducing the toxicity of the existing chemotherapeutic drug is a challenge for us for safeguarding the human beings from cancer. There are evidences that metronomic chemotherapy and combination of various chemotherapeutic drugs can enhance the efficacy of chemotherapeutic drugs. So, intensive research regarding the metronomic chemotherapy and the combination chemotherapy is needed. Besides, search for new drug should also be continued.
Abbreviations
MTX: Methotrexate, IUPAC: International Union of Pure and Applied Chemistry, RA: Rheumatoid Arthritis, GVHD: Graft-Vs.-Host Disease, CSP: Cyclosporine, DHFR: Dihydrofolate Reductase, TS: Thymidylate Synthase, dUMP: Deoxyuridine Monophosphate, dTMP: Deoxythymidine Monophosphate, CP: Cyclophosphamide, DNA: Deoxyribonucleotide Acid, 5-FU: 5-Fluorouracil, RNA: Ribonucleic Acid, FR: Free Radical, ROS: Reactive Oxygen Species, AP: Activator Protein, NF: Nuclear Factor, TRE: TPA-response Element, MAP: Mitogen-activated Protein, AK-2: Adenylate Kinase-2, Smac: Second mitochondria-derived activator of caspases, AIF: Apoptosis inducing Factor, Apaf- 1: Apoptotic Protease Activating Factor-1, PT: Permeability Transition, ATP: Adenosine Triphosphate, CARD: Caspase Recruitment Domain, ADP: Adenosine Diphosphate, PAC-1: First Procaspase Activating Compound, PAK2: p21 Activated Kinase 2, CAD: Caspase-activated DNase, ICAD: Inhibitor of CAD, DFF: DNA Fragmentation Factor, Lys: Lysine, Ser: Serine, GSH: Glutathione, 4-HC: 4- hydroxycyclophosphamide, FUTP: Fluorouridine Triphosphate, FdUMP: Fluorodeoxyuridine Monophosphate, FdUTP: Fluorodeoxyuridine Triphosphate, FUMP: Fluorouridine Monophosphate, OPRT: OrotatePhosphoribosyltransferase, PRPP: Phosphoribosyl Pyrophosphate, FUR: Fluorouridine, UP: UridinePhosphorylase, UK: Uridine Kinase, FUDP: FluorouridineDiphosphate, snRNA: Small Nuclear Ribonucleic Acid, FdUDP: FluorodeoxyuridineDiphosphate, RR: RibonucleotideReductase, FUDR: Fluorodeoxyuridine, TP: Thymidine Phosphorylase, TK: Thymidine Kinase, THF: Tetrahydrofolate, dTTP: DeoxythymidineTriophosphate, dATP: Deoxyadenosine Triphosphate, dGTP: Deoxyguanosine Triphosphate, dCTP: Deoxycytidine Triphosphate, dUTP: Deoxyuridine Triphosphate, dNTP: Deoxynucleotide, UDG: Uracil-DNA-Glycosylase.
References
-
N.K.
Agarwal,
G.A.
Mueller,
C.
Mueller,
J.H.
Streich,
A.R.
Asif,
H.
Dihazi.
Expression proteomics of acute promyelocytic leukaemia cells treated with methotrexate. Biochim Biophys Acta.
2010;
1804
:
918-928
.
-
Airley.
Cancer chemotherapy: basic science to the clinic. Wiley-Blackwell, West Sussex.
2009
.
-
F.J.
Alonso,
J.A.
Segura,
J.
Lora,
C.
Lobo,
B.
Fernandez-Molina,
J.
Marquez,
J.M.
Mates.
Sensitisation of Ehrlich ascitic tumour cells to methotrexate by inhibiting glutaminase. Anticancer research.
2005;
25
:
3315-3320
.
-
W.S.
Arthur Camerman.
Activated cyclophosphamide anticancer drugs: molecular structure of 4-hydroperoxycyclophosphamide. Acta Cryst B.
1977;
33
:
678-683
.
-
T.
Brummaier,
E.
Pohanka,
A.
Studnicka-Benke,
H.
Pieringer.
Using cyclophosphamide in inflammatory rheumatic diseases. Eur J Intern Med.
2013;
24
:
590-596
.
-
L.
Cai,
B.F.
Hales,
B.
Robaire.
Induction of apoptosis in the germ cells of adult male rats after exposure to cyclophosphamide. Biol Reprod.
1997;
56
:
1490-1497
.
-
G.
Cao,
W.
Pei,
J.
Lan,
R.A.
Stetler,
Y.
Luo,
T.
Nagayama,
S.H.
Graham,
X.M.
Yin,
R.P.
Simon,
J.
Chen.
Caspaseactivated DNase/DNA fragmentation factor 40 mediates apoptotic DNA fragmentation in transient cerebral ischemia and in neuronal cultures. J Neurosci.
2001;
21
:
4678-4690
.
-
C.-S.
Chen,
J.T.
Lin,
K.A.
Goss,
Y.-a.
He,
J.R.
Halpert,
D.J.
Waxman.
Activation of the anticancer prodrugs cyclophosphamide and ifosfamide: identification of cytochrome P450 2B enzymes and site-specific mutants with improved enzyme kinetics. Mol Pharmacol.
2004;
65
:
1278-1285
.
-
Y.-X.
Chen,
W.-G.
Lv,
H.-Z.
Chen,
F.
Ye,
X.
Xie.
Methotrexate induces apoptosis of human choriocarcinoma cell line JAR via a mitochondrial pathway. Eur J Obstet Gynecol Reprod Biol.
2009;
143
:
107-111
.
-
Y.W.
Chung,
D.W.
Jeong,
J.Y.
Won,
E.J.
Choi,
Y.H.
Choi,
I.Y.
Kim.
H(2)O(2)-induced AP-1 activation and its effect on p21(WAF1/CIP1)-mediated G2/M arrest in a p53-deficient human lung cancer cell. Biochem Biophys Res Commun.
2002;
293
:
1248-1253
.
-
L.
Clarke,
D.J.
Waxman.
Oxidative metabolism of cyclophosphamide: identification of the hepatic monooxygenase catalysts of drug activation. Cancer Res.
1989;
49
:
2344-2350
.
-
B.N.
Cronstein.
Low-dose methotrexate: a mainstay in the treatment of rheumatoid arthritis. Pharmacol Rev.
2005;
57
:
163-172
.
-
A.A.
Dayem,
H.Y.
Choi,
J.H.
Kim,
S.G.
Cho.
Role of oxidative stress in stem, cancer, and cancer stem cells. Cancers (Basel).
2010;
2
:
859-884
.
-
L.
Genestier,
R.
Paillot,
S.
Fournel,
C.
Ferraro,
P.
Miossec,
J.P.
Revillard.
Immunosuppressive properties of methotrexate: apoptosis and clonal deletion of activated peripheral T cells. J Clin Invest.
1998;
102
:
322-328
.
-
D.
Hanahan,
R.A.
Weinberg.
Hallmarks of cancer: the next generation. Cell.
2011;
144
:
646-674
.
-
Haubitz.
Acute and long-term toxicity of cyclophosphamide. Tx Med.
2007;
19
:
26-31
.
-
W.Y.
Huang,
P.M.
Yang,
Y.F.
Chang,
V.E.
Marquez,
C.C.
Chen.
Methotrexate induces apoptosis through p53/p21-dependent pathway and increases E-cadherin expression through downregulation of HDAC/EZH2. Biochemical pharmacology.
2011;
81
:
510-517
.
-
R.U.
Janicke,
P.
Ng,
M.L.
Sprengart,
A.G.
Porter.
Caspase-3 is required for alpha-fodrin cleavage but dispensable for cleavage of other death substrates in apoptosis. J Biol Chem.
1998;
273
:
1554015545
.
-
F.
Juma,
T.
Ogada.
Pharmacokinetics of cyclophosphamide in Kenyan Africans. British journal of clinical pharmacology.
1983;
16
:
61-63
.
-
J.
Kaysen,
D.
Spriggs,
D.
Kufe.
Incorporation of 5- fluorodeoxycytidine and metabolites into nucleic acids of human MCF-7 breast carcinoma cells. Cancer Res.
1986;
46
:
4534-4538
.
-
R. C.
Koya,
H.
Fujita,
S.
Shimizu,
M.
Ohtsu,
M.
Takimoto,
Y.
Tsujimoto,
N.
Kuzumaki.
Gelsolin inhibits apoptosis by blocking mitochondrial membrane potential loss and cytochrome c release. J Biol Chem.
2000;
275
:
15343-15349
.
-
P.
Kozub,
M.
Simaljakova.
Systemic therapy of psoriasis: methotrexate. Bratisl Lek Listy.
2011;
112
:
390-394
.
-
M.
Lamberti,
S.
Porto,
M.
Marra,
S.
Zappavigna,
A.
Grimaldi,
D.
Feola,
D.
Pesce,
S.
Naviglio,
A.
Spina,
N.
Sannolo.
5-Fluorouracil induces apoptosis in rat cardiocytes through intracellular oxidative stress. J Exp Clin Cancer Res.
2012;
31
:
60-60
.
-
A.
Lawen.
Apoptosis-an introduction. Bioessays.
2003;
25
:
888-896
.
-
M.C.
Li.
Current status of cancer chemotherapy. J Natl Med Assoc.
1960;
52
:
315-320
.
-
D.B.
Longley,
D.P.
Harkin,
P.G.
Johnston.
5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer.
2003;
3
:
330-338
.
-
S.
Louie,
G.A.
Lillington.
Low dose methotrexate pneumonitis in rheumatoid arthritis. Thorax.
1986;
41
:
703-704
.
-
J.D.
Ly,
D.R.
Grubb,
A.
Lawen.
The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis.
2003;
8
:
115-128
.
-
M.
Malet-Martino,
R.
Martino.
Clinical studies of three oral prodrugs of 5-fluorouracil (capecitabine, UFT, S-1): a review. Oncologist.
2002;
7
:
288-323
.
-
D.
McIlroy,
M.
Tanaka,
H.
Sakahira,
H.
Fukuyama,
M.
Suzuki,
K.
Yamamura,
Y.
Ohsawa,
Y.
Uchiyama,
S.
Nagata.
An auxiliary mode of apoptotic DNA fragmentation provided by phagocytes. Genes & development.
2000;
14
:
549-558
.
-
L.
Mojardin,
J.
Botet,
L.
Quintales,
S.
Moreno,
M.
Salas.
New insights into the RNA-based mechanism of action of the anticancer drug 5’-fluorouracil in eukaryotic cells. PLoS One.
2013;
8
:
e78172
.
-
M.M.
Morais,
J.N.
Belarmino-Filho,
G.A.
Brito,
R.A.
Ribeiro.
Pharmacological and histopathological study of cyclophosphamide-induced hemorrhagic cystitis - comparison of the effects of dexamethasone and Mesna. Braz J Med Biol Res.
1999;
32
:
1211-1215
.
-
N.
O’Donovan,
J.
Crown,
H.
Stunell,
A.D.
Hill,
E.
McDermott,
N.
O’Higgins,
M.J.
Duffy.
Caspase 3 in breast cancer. Clin Cancer Res.
2003;
9
:
738-742
.
-
M.
Ott,
J.D.
Robertson,
V.
Gogvadze,
B.
Zhivotovsky,
S.
Orrenius.
Cytochrome c release from mitochondria proceeds by a two- step process. Proc Natl Acad Sci U S A.
2002;
99
:
1259-1263
.
-
K. V. M.A.
Pavan,
Ravikiran
A.,
Kamaraj
P..
Sorption- Desorption Behavior and Characterization of Cyclophosphamide. Chem Sci Trans.
2013;
2
:
135-140
.
-
M.S.
Pendekal,
P.K.
Tegginamat.
Development and characterization of chitosan-polycarbophil interpolyelectrolyte complexbased 5-fluorouracil formulations for buccal, vaginal and rectal application. Daru.
2012;
20
:
67
.
-
Q.P.
Peterson,
D.R.
Goode,
D.C.
West,
K.N.
Ramsey,
J.J.Y.
Lee,
P.J.
Hergenrother.
PAC-1 activates procaspase-3 in vitro through relief of zinc-mediated inhibition. J Mol Biol.
2009;
388
:
144-158
.
-
G.V.
Putcha,
C.A.
Harris,
K.L.
Moulder,
R.M.
Easton,
C.B.
Thompson,
E.M. Jr.
Johnson.
Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J Cell Biol.
2002;
157
:
441-453
.
-
H.Y.
Rahiem Ahmed YAA.
Prevention and management of high dose methotrexate toxicity. J Cancer Sci Ther.
2013;
5
:
106-112
.
-
C.
Roobol,
G.B.
De Dobbeleer,
J.L.
Bernheim.
5- fluorouracil and 5-fluoro-2’-deoxyuridine follow different metabolic pathways in the induction of cell lethality in L1210 leukaemia. Br J Cancer.
1984;
49
:
739-744
.
-
M.
Ross,
G.M.
Schmidt,
J.C.
Niland,
M.D.
Amylon,
A.C.
Dagis,
G.D.
Long,
A.P.
Nademanee,
R.S.
Negrin,
M.R.
O’Donnell,
P.M.
Parker.
Cyclosporine, methotrexate, and prednisone compared with cyclosporine and prednisone for prevention of acute graftvs.-host disease: effect on chronic graft-vs.-host disease and long-term survival. Biology of blood and marrow transplantation: journal of the American Society for Blood and Marrow Transplantation.
1999;
5
:
285-291
.
-
Abdulla M.d.A.
Saleh,
Cheung
Bernard.
Methotrexate in psoriasis: from A to Z. J Turk Acad Dermatol.
2010;
4
:
1-13
.
-
Ajit. K
Saxena,
Singh
Gajendra.
Structural interaction between drug - DNA and protein- A novel approach for bioinformatics in medicine. Biomedical Research.
2009;
20
:
28-34
.
-
M.
Schuler,
D.R.
Green.
Mechanisms of p53-dependent apoptosis. Biochem Soc Trans.
2001;
29
:
684-688
.
-
P.S.
Schwartz,
D.J.
Waxman.
Cyclophosphamide induces caspase 9-dependent apoptosis in 9 L tumor cells. Mol Pharmacol.
2001;
60
:
12681279
.
-
C.K.
Sen,
L.
Packer.
Antioxidant and redox regulation of gene transcription. FASEB journal: official publication of the Federation of American Societies for Experimental Biology.
1996;
10
:
709-720
.
-
A.J.
Steele,
A.G.
Prentice,
A.V.
Hoffbrand,
B.C.
Yogashangary,
S.M.
Hart,
E.P.
Nacheva,
J.D.
Howard-Reeves,
V.M.
Duke,
P.D.
Kottaridis,
K.
Cwynarski.
p53-mediated apoptosis of CLL cells: evidence for a transcription-in dependent mechanism. Blood.
2008;
112
:
38273834
.
-
F.
Steger,
M.G.
Hautmann,
O.
Kolbl.
5-FU-induced cardiac toxicity-an underestimated problem in radio oncology?. Radiat Oncol.
2012;
7
:
212
.
-
R.
Storb,
H.J.
Deeg,
M.
Pepe,
F.
Appelbaum,
C.
Anasetti,
P.
Beatty,
W.
Bensinger,
R.
Berenson,
C.D.
Buckner,
R.
Clift.
Methotrexate and cyclosporine versus cyclosporine alone for prophylaxis of graft-versus-host disease in patients given HLA-identical marrow grafts for leukemia: long-term follow-up of a controlled trial. Blood.
1989;
73
:
17291734
.
-
S.
Tariq,
S.M.
Tariq.
Methotrexate in rheumatoid arthritis: can current knowledge and experience justify its use as a first-line disease- modifying agent?. Postgrad Med J.
1993;
69
:
775-780
.
-
L.B.
Travis,
R.E.
Curtis,
B.
Glimelius,
E.J.
Holowaty,
F.E.
Van Leeuwen,
C.F.
Lynch,
A.
Hagenbeek,
M.
Stovall,
P.M.
Banks,
J.
Adami.
Bladder and kidney cancer following cyclophosphamide therapy for non-Hodgkin’s lymphoma. J Natl Cancer Inst.
1995;
87
:
524-530
.
-
M.
Tsai-Turton,
B.T.
Luong,
Y.
Tan,
U.
Luderer.
Cyclophosphamide-induced apoptosis in COV434 human granulosa cells involves oxidative stress and glutathione depletion. Toxicol Sci.
2007;
98
:
216230
.
-
H.
Ungefroren,
S.
Sebens,
D.
Seidl,
H.
Lehnert,
R.
Hass.
Interaction of tumor cells with the microenvironment. Cell Communication and Signaling: CCS.
2011;
9
:
18-18
.
-
B.
Van Triest,
H.M.
Pinedo,
G.
Giaccone,
G.J.
Peters.
Downstream molecular determinants of response to 5-fluorouracil and antifolate thymidylate synthase inhibitors. Ann Oncol.
2000;
11
:
385-391
.
-
A.
Bueltzingsloewen,
R.
Belanger,
C.
Perreault,
Y.
Bonny,
D.C.
Roy,
Y.
Lalonde,
J.
Boileau,
J.
Kassis,
R.
Lavallee,
M.
Lacombe.
Acute graft-versus-host disease prophylaxis with methotrexate and cyclosporine after busulfan and cyclophosphamide in patients with hematologic malignancies. Blood.
1993;
81
:
849-855
.
-
C.-C.L.
Wang Tso-Fu,
Chao-Yuan Yao
Sung-Chao Chu.
Treatment of metastatic or recurrent gastric cancer with weekly 24-hour infusion of cisplatin and high-dose 5-fluorouracil/leucovorin in an outpatient setting. Tzu Chi Med J.
2006;
18
:
432-437
.
-
Y.N.
Yoshikawa Toshikazu.
What is oxidative stress?. JMAJ.
2002;
45
:
271-276
.
-
X.
Zhao,
Y.T.
Yu.
Incorporation of 5-fluorouracil into U2 snRNA blocks pseudouridylation and pre-mRNA splicing in vivo. Nucleic Acids Res.
2007;
35
:
550-558
.
Comments
Downloads
Article Details
Volume & Issue : Vol 2 No 02 (2015)
Page No.: 196-206
Published on: 2015-02-19
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 8150 times
- Download PDF downloaded - 2550 times
- View Article downloaded - 8 times

{kind=link}
{kind=link}
{kind=link}
{kind=link}