

Constitutive Photomorphogensis Protein1 (COP1) mediated p53 pathway and its oncogenic role
Copyrights: Md. Golam Rabbani, Sk Amir Hossain, Khandker Khaldun Islam, Sarder Nasir Uddin, 2014. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
We have reviewed the COP1 mediated tumor suppressor protein p53 pathway and its oncogenic role. COP1 is a negative regulator of p53 and acts as a pivotal controller of p53-Akt death-live switch (Protein kinase B). In presence of p53, COP1 is overexpressed in breast, ovarian, gastric cancers, even without MDM2 (Mouse double minute-2) amplification. Following DNA damage, COP1 is phosphorylated instantly by ATM (Ataxia telangiectasia mutated) and degraded by 14-3-3σ following nuclear export and enhancing ubiquitination. In ATM lacking cell, other kinases, i.e. ATR (ataxia telangiectasia and Rad3-related protein), Jun kinases and DNA-PK (DNA-dependent protein kinase) cause COP1 & CSN3 (COP9 signalosome complex subunit-3) phosphorylation and initiate COP1’s down regulation. Although, it has been previously found that co-knockout of MDM2 and COP1 enhance p53’s half life by eight fold, the reason is still unknown. Additionally, while interacting with p53, COP1 upregulate MDM2’s E3 ubiquitin ligase, Akt, CSN6 (COP9 signalosome 6) activity and inhibit 14-3-3σ’s negative regulation on MDM2 and COP1 itself. Conclusively, there persists an amplification loop among COP1, MDM2, Akt and 14-3-3σ to regulate p53’s stability and activity. However, the role of another tumor suppressor PTEN (phosphatase and tensin homologue) is yet to be discovered. This study provides insight on the molecular genetic pathways related to cancer and might be helpful for therapeutic inventions.
Introduction
In plant, COP1 is a crucial mediator to block Photomorphogenesis during dark through ubiquitin mediated proteasomal degradation of light-induced transcription factor Hypocotyl 5 (HY5) Hardtke et al., 2000. Human constitutive photomorphogensis protein 1 (huCOP1= “COP1”) E3 ubiquitin ligase, is reportedly overexpressed in breast, ovarian cancer Dornan et al., 2004a, and hepatocellular carcinomas (HCC), third most lethal neoplasm Lee et al., 2010 and gastric cancer and predictive of survival in the latter Li et al., 2012. Therefore, it contributes to accelerate degradation of the p53 protein also attenuating p53’s function Dornan et al., 2004b. The p53 is dubbed as “the guardian of genome” Lane, 1992 which reflects its real function. It is the single hub through which various kind of signals pass and expresses decisive cellular response Lu, 2010. The p53 applies emergency break when DNA is damaged by irradiation, mutagenic chemicals and other stress conditions. Following DNA damage, damage sensing molecules such as ATM becomes active and phosphorylates p53 such as at serine-(ser)-15 Su et al., 2010. The p53 becomes an active transcription factor and induces transcription of several genes to mediate cell cycle arrest, DNA repair, apoptosis such as p21, 14-3-3σ (sigma), PTEN and inhibit E3 ligases such as MDM2, COP1, p53-induced protein with a RING-h2 domain (Pirh2) Dornan et al., 2004b. E3 ligases inhibit the function of p53 by causing its ubiquitin mediated proteolysis. p53-Akt act as death-live Switch. Death-live Switch becomes on/off depending on Bistability. In respect to p53-Akt death-live Switch, p53 active means the immediate pause of cell growth or apoptosis and Akt active means the cell will proceed on Wee and Aguda,2006. COP1 is the pivotal molecule of the p53 and 14-3-3σ, p53 and MDM2 & Akt and MDM2 pathways. While COP1 increases, 14-3-3σ decreases and Akt activates; and thus COP1 prevents p53 dependent apoptosis and induces cell growth.
COP1 regulates various cellular functions, for instance, proliferation and survival, through ubiquitinmediated protein degradation. Ubiquitinated target of COP1, such as p53, is the first identified target of COP1 in mammal Dornan et al., 2004b. COP1 is able to ubiquitinate p53 without MDM2 or Pirh2 Bianchi et al., 2003. Silencing of COP1 and MDM2 enhances half-life of p53 two fold in comparative to control respectively. But co-silencing of MDM2 and COP1 enhance half-life of p53 eight-fold in relative to control Dornan et al., 2004b. This effect is double than the summation of MDM2 and COP1’s independent effects. Therefore, COP1 axis plays pivotal role in spreading cancer.
Regulation of cop1 through atm
ATM protein kinase, one of the critical components of DNA damage sensing molecule, protects genomic integrity. ATM is a molecular police for DNA damage and phosphorylates key effecter substrates like the tumor suppressor p53 and E3 ligases such COP1, MDM2 to dampen their ability to negatively regulate p53 Pereg et al.,2005. These three enzymes: ATM, ATR and DNA-PK are involved in DNA damage responses. ATM and ATR participate in the activation of cell-cycle checkpoints induced by DNA damage (Keith and Schreiber, 1995), whereas DNA-PK acts primarily during DNA repair. The ATM protein is encoded by the ATM gene responsible for the human genetic disorder Ataxia telangiectasia (AT), a rare autosomal recessive genetic disorder Anderson and Carter, 1996.
ATM – p53
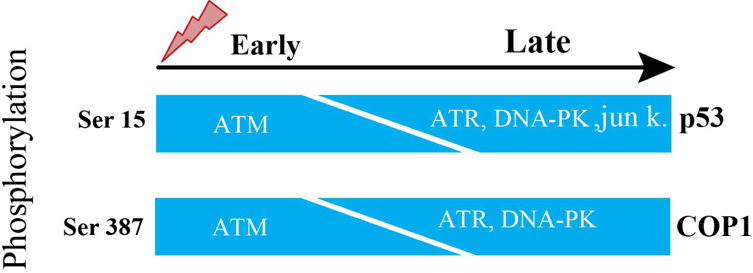
The p53 is tightly regulated by controlled degradation through E3 ubiquitin ligases such as COP1, is a critical negative regulator of p53 Dornan et al., 2004b. Therefore, in response to DNA damage, ATM phosphorylates Ser387 on COP1, which is necessary and suffisufficient to disrupt the COP1-p53 complex and subsequently to abrogate the ubiquitination and degradation of p53 thus p53 becomes stabilized and exerts its tumor suppressor properties in response to DNA damage Dornan et al., 2006. Activation of ATM causes decrease in the abundance of COP1 protein. Ubiquitination event entirely depends on phosphorylation at Ser-387 on COP1. Ser-15 is a functionally important residue within the p53 amino-terminal region. Ser-15 on p53, phosphorylated by ATM, represents an early cellular response to a variety of genotoxic stresses Siliciano et al.,1997. Therefore, ATM is directly responsible for phosphorylation and degradation of COP1.
ATM lacking fibroblasts
The steady-state levels of COP1 in human fibroblasts after DNA damage induced by 10 Geiger (Gy) of IR started to decline within 30 minutes and decreased to an almost undetectable level by 1 hour after exposure to IR. This is closely correlated to an increase in steady-state levels of p53 and activation of the downstream target genep21. There is decrease of COP1 protein abundance but not decrease of COP1 in mRNA levels, besides same result with as low as 2 Gy of IR. Human fibroblasts, derived from A-T patient, lack the function of ATM. When these fibroblasts were stimulated by 10 Gy IR, steady-state level of COP1 protein was unchanged in these fibroblasts at early time points but decreased in abundance at later time points Dornan et al., 2006. From this above information, it is clear that there presents some other molecules which can subsidize the lack of ATM. ATR functions as an upstream regulator of p53 phosphorylation in DNAdamaged cells. Both ATM and ATR play partially overlapping and independent roles in the phosphorylation of p53 during cellular exposure to genotoxic stress. In fact, ATM’s catalytic activity is more robust and quicker than ATR’s catalytic activity. Consequently, when ATM’s activation is abrogated, ATR phosphorylates Ser-15 on p53 but takes longer time due to ATR’s slower catalytic activity than ATM Tibbetts et al., 1999, which is consistent with the result of Dornan et al., 2006 report. In response to DNA damage, Ser15 on p53 is reported to be phosphorylated by DNA-PK and Jun kinases at later inductive phase Lees-Miller et al.,1992. ATM phosphorylates ser-15 on p53 and ser-387 on COP1 at the same time.
Therefore, it could be fairly proposed that these three candidates of kinases named ATR, DNA-PK and Jun kinases can phosphorylate ser-387 on COP1 and initiate degradation of COP1 in ATM lacking cells ( Figure 1 ).
Human cop1
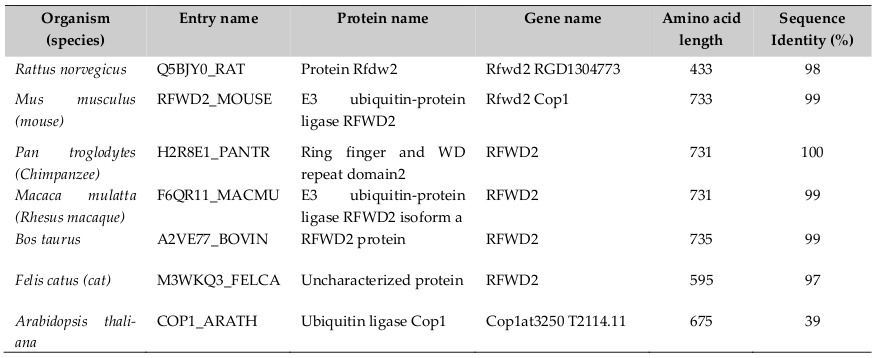
Human COP1 officially known as Ring finger and WD repeat domain 2 (RFND2); synonyms are- COP1 and RNF200, locates on chromosome 1q25.1-q25.2. It consists of 21 exons and span 263kb and encodes four isoforms (www.uniprot.org). Its sequence length is 2193 nucleotides, encoding protein sequence length 731 amino acids (a. a). It is found in plants, lower animal and human beings. Therefore, COP1 is evolutionary well conserved across species ( Table 1 ). Dornan et al., 2004a reported that the COP1 gene contains two p53 consensus recognition sites (PuPuPuCWWGPyPyPy) (Pu= purine and Py= pyrimidine) on the COP1 promoter at -2074 to -2083 and- 2086 to-2095 from +1 (ATG) transcription start point. Therefore, COP1 is a p53’s transcriptional target gene. It also reported that p53 is the first identified direct mammalian substrate of COP1 which binds p53 protein through the 92-160 and 318-393 Amino acid sequences Yamada et al., 2013. Thereby, there exists a negative feedback loop between COP1 and p53.
Function of COP1 Domains
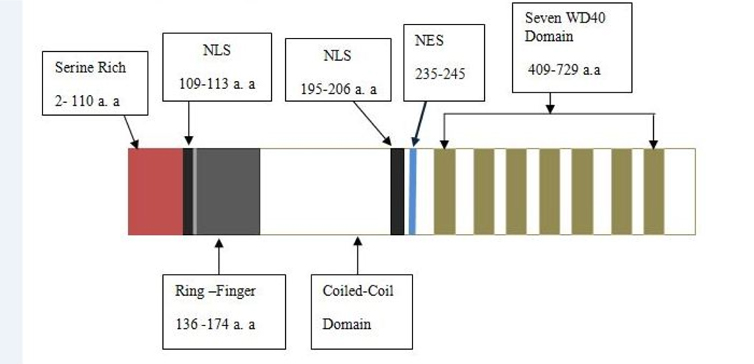
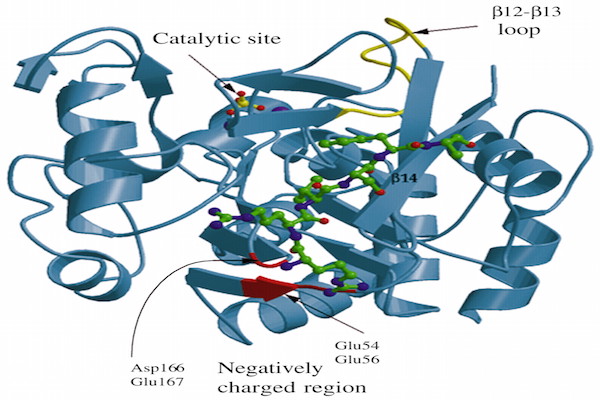
COP1 contains three domains- Ring, Coiled-coil and WD40. COP1 also contains two nuclear localization sequences (NLS) and one nuclear export sequence (NES). Coiled-coil domain locates in between Ring and WD40 domains. The Ring finger domain is marked by conserved pattern of cysteine and histidine residues that involves binding two zinc atoms in a particular cross-brace orientation (Von Arnim and Deng, 1993). Ring domain involves Ring or non-Ring protein-protein interaction. The Ring finger domain is necessary for COP1’s E3 ubiquitin ligase activity. Coiled-coil domain is important for COP1’s selfdimerization. A monopatite NLS locates in between amino-terminal and Ring domain and a bipartite NLS is located proximal at the WD40 repeats ( Figure 2 ). COP1 localizes both in cytoplasm and nucleus. In unstressed condition, COP1 is mainly localized in the nucleus of cell Dornan et al., 2006.
COP1’s E3 ligases effect
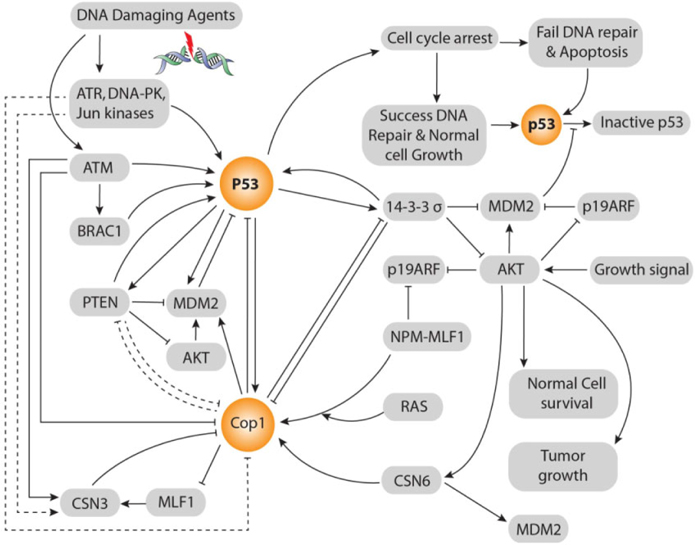
According to Dornan et al. Dornan et al., 2004b, COP1’s negative effect on p53 is 2-fold in respect to control also MDM2 and Pirh2’s negative effect on p53 is 2-fold and 1.5-fold respectively. Co-ablation of COP1 and Pirh2 gives 3.5-fold negative effects on p53 that is equal to the summation of these two E3 ubiquitin ligases independent negative effects on p53. Whereas, co-ablation of MDM2 and COP1 gives 8-fold negative effects on p53 that is double to the summation of these two E3 ubiquitin ligases independent negative effects on p53. Although ligase activity of COP1 and MDM2 is independent, co-ablation of MDM2 and COP1 gives 8-fold negative effects on p53. There is evidence that COP1 is located on upstream of Akt and MDM2 and parallel to 14-3-3σ and on downstream of p53 in ATM and MLF1 mediated pathways Yoneda-Kato et al., 2005. Up-stream relationship between COP1 and 14-3-3σ is vice versa. But, 14-3-3σ is always located in up-stream of MDM2. COP1 locates in the middle of p53 and Akt signaling pathways. Thereby, besides COP1’s independent E3 ubiquitin ligases effect on p53, COP1 enhances MDM2’s E3 Ubiquitin ligase activity through direct interaction, by enhancing Akt activity also inhibiting 14-3-3σ’s negative regulation on MDM2. Thus inhibiting 14-3-3σ’s activity, COP1 itself reduces the 14-3-3σ inhibition upon COP1. Hence, there persists a cascade between COP1 and MDM2 axis that vigorously control p53’s stability and activity ( Figure 3 ).
14-3-3 σ Protein
14-3-3σ is originally isolated from human mammary epithelial cells and known as stratifin (SFN) due to its expression in stratified epithelia. It has two unique residues at Ser5 and Glutamine (Glut)-80, which lie opposite to each other. On the contrary, other 14-3-3 isoforms have Asp 5 or Glu 5 residues. Because of these two unique residues the σ isoform cannot form heterodimer with other isoforms. The σ isoform is responsible for binding particular targets, which are not recognized by other members of 14-3-3 proteins family (Wilker et al., 2005).
p53-14-3-3σ Positive Feedback Loop
14-3-3σ gene locates in chromosome 1p35 and its promoter contains a p53 response element; therefore, p53 can directly transactivate the expression of 14-3-3σ following DNA damage Hermeking et al., 1997. 14-3- 3σ binds and activates p53 in a positive feedback loop. It also acts as a negative regulator of Akt Yang et al., 2007Yang et al.,2006. It is down regulated during neoplastic transformation such as breast cancer, HCC, ovarian cancer Iwata et al., 2000. 14-3-3σ’s roles in p53 tetramerization which is critical for both p53’s binding to DNA sequence and facilitating p53’s transcriptional activity Waterman et al., 1998. Thus, 14-3- 3σ positively regulates p53 protein through stabilizing p53 and increasing p53 transcriptional activity.
14-3-3σ is induced by p53 in response to DNA damage
p53 induces expression of 14-3-3σ to mediate cell cycle arrest. 14-3-3σ functions as a secondary level of tumor suppressor. In the absence of 14-3-3σ, cyclinB/cdc2 complexes enter the nucleus even existed DNA damage, causing mitotic catastrophe, a situation in which mitosis is induced with DNA damage (Chan et al., 1999). Lack of 14-3-3σ’s activation causes immortality of cells, leads to tumor formation. Overexpression of 14-3-3σ suppresses the anchorage-independent growth of several breast cancer cell lines (Laronga et al., 2000). Hence, the tumor suppressor function of 14- 3-3σ is abolished during tumorigenesis. 14-3-3σ upregulation correlates with Akt inactivation in response to DNA damage. There exists a strong inverse relationship between 14-3-3σ expression and the activation of Akt. 14-3-3σ cannot function as a tumor suppressor during tumorigenesis due to low expression or silencing Ferguson et al., 2000.
14 -3-3σ has a Positive Feedback Effect on p53
The expression level of p53 is increased when a high level of 14-3-3σ is present; as a result, 14-3-3σ has a positive impact on p53 stability. Elevated level of 14-3- 3σ mediates p53’s stabilization directly by increasing p53’s half-life, while elevated level of 14-3-3σ leads to a decrease in the half-life of MDM2 and COP1.14-3-3σ directly inhibits MDM2-mediated p53’s ubiquitination, nuclear export and degradation by the cytoplasmic proteosome. 14-3-3σ blocks MDM2-mediated nuclear export of p53 very efficiently, thereby stabilizing and retaining p53 in the nucleus Yang et al., 2003.
14-3-3σ -MDM2
MDM2 gene locates on chromosome12q13. MDM2 contains both NLS and NES. Therefore, MDM2 moves between the cytoplasm and nucleus. Akt phosphorylates MDM2 on Ser-166 and Ser-188 thus inhibiting MDM2’s self-ubiquitination (Feng et al., 2004) and induces localization of MDM2 into the nucleus. Through the binding of MDM2 with p53, it promotes p53’s nuclear export and cytoplasmic degradation (Zhou et al., 2001). 14-3-3σ blocks MDM2-mediated nuclear export, thereby stabilizes and retains p53 in the nucleus Yang et al., 2007. In conclusion, Akt-meditaed cell survival is inhibited by 14-3-3σ.
AKT-PTEN
Protein kinase B (PKB, also called Akt) oncogene is a crucial regulator of a variety of cellular processes, including cell survival and proliferation. Akt activity is elevated in several types of human malignancy, including ovarian, breast, lung, and thyroid cancers Vivanco and Sawyers, 2002. The kinase activity of Akt is activated in human cancer as a result of dysregulation of its regulators such as PTEN. It, located on chromosome 10, is a transcriptional target of p53. It can inhibit Akt; thereby affecting the subcellular localization of MDM2. It can down regulate MDM2 and increase p53’s stability Freeman et al., 2003. In addition, PTEN-/- cells have high Akt activity and are defective in checkpoint control in response to DNA damage Puc et al., 2005. As a result, there exists positive feedback loop between p53 and PTEN. Significantly, 14-3-3σ and PTEN impose same biological effects in terms of p53 activation. 14-3-3σ inhibits E3 ligase activity of MDM2, Akt and COP1. On the contrary, COP1 negatively regulates 14-3-3σ. Similarly, PTEN negatively regulates MDM2 and Akt Freeman et al., 2003 but lacks the interaction information with COP1. Therefore, 14-3-3σ and PTEN play tumor suppressive role in the same way. Thereby, there presents vice versa relationship between PTEN and COP1 ( Figure 3 ).
Myeloid leukemia factor 1 (mlf1)
MLF1 is generated by t (3; 5) (q25.1; q34) chromosomal translocation, which is associated with myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) Yoneda-Kato et al., 1996. Human MLF1 has a single nucleotide polymorphism at the 226th aminoacid codon, which produces proline (P) and threonine (T) at a ratio of 3:1 in normal genomic alleles Fukumoto et al., 2005. Cells expressing T-MLF1 cease proliferation much earlier than those expressing PMLF1 like as ATM’s response to DNA damage signal.
Consensus Sequence of MLF1
MLF1 continuously shuttles between the nucleus and the cytoplasm of proliferating mammalian cells by interacting directly with Chromosomal Region Maintenance-1 (CRM1, also known as Exportin-1). MLF1 contains a functional NES sequence at amino acids 89 to 96, which enables binding of CRM1 in vivo Fukuda et al., 1997. Two putative consensus sequences of NLS located at 168 to 174 & 232 to 236 sequences are conserved in human and mouse MLF1 Yoneda-Kato et al., 2005.
Physiological Function of MLF1
MLF1, a negative regulator of cell cycle progression, is physiologically involved in tumor suppressor pathways that functions up-stream of the tumor suppressor p53 Dornan et al., 2004b. Genotoxic stress signals and the MLF1 signal are capable of inducing the CSN3-COP1 mediated accumulation of p53. MLF1 stabilizes p53’s activity by suppressing COP1, through a third component CSN3. It directly binds CSN3 and activates the p53/p21 pathway through CSN3–COP1 pathway. MLF1’s Residues from 50 to 125 are essential for interaction with CSN3. More than average expression of CSN3 is essential for the growth suppressive function of MLF1 Yoneda-Kato and Kato, 2008Yoneda-Kato et al., 2005. CSN3 expression causes decrease of COP1 protein in the endogenous. On the other hand, a reduction of CSN3 leads to an increase of COP1 regardless the level of MLF1 expression. Hence, MLF1 modulates COP1 expression through CSN3 to activate the p53 pathway. Interestingly, CSN3 is a substrate of ATM in DNA damage-induced apoptosis Lavin and Kozlov, 2007. Therefore, CSN3 might be a substrate of ATR, DNA-PK and Jun kinase in ATM lacking cells like as COP1 ser387 ( Figure 1 ).
Csn6, a multiprotein complex
CSN6 consists of eight distinct subunits (CSN1-CSN8) in all eukaryotes. Akt phosphorylates at ser-60 to stabilize CSN6. Interestingly, CSN6 activates Akt to create a self-propelling positive feed-back loop Chen et al., 2012. CSN6 directly ties COP1and increases COP1’s stability through inhibition of ubiquitinmediated COP1’s proteasomal degradation. Thereby, CSN6 up-regulates COP1 at the post-transcriptional level. CSN6 regulates 14-3-3σ’s post-transcriptional level by enhancing 14-3-3σ’s ubiquitination through COP1 ( Figure 3 ).
Down-regulation of 14-3-3σ by CSN6 is independent on p53 expression. CSN6 forms tie with the Cterminus of 14-3-3σ by N-terminus of CSN6. Thus, it can down regulate the expression of 14-3-3σ and lead Akt activation. In this way, both CSN6 and COP1 are involved in degrading 14-3-3σ, promoting cell survival and increasing tumorigenicity. 14-3-3σ is down regulated during neoplastic transformation Prasad et al., 1992. On the contrary, 14-3-3σ negatively regulates COP1. Therefore, COP1and 14-3-3σ form a vice versa relationship between them. CSN6 is a positive regulator of MDM2 Lee et al., 2011. In a word, CSN6 destabilizes p53 to promote tumorigenesis ( Figure 3 ).
Npm-Mlf1
The fusion protein NPM-MLF1, generated by t(3; 5) (q25.1; q34) chromosomal translocation, leads to leukemogenesis and it consists of more than half of the amino terminus of NPM and almost the entire MLF1 sequence Yoneda-Kato et al., 1996. The region of NPM retained in NPM-MLF1 includes a NES sequence and a bipartite NLS sequence Wang et al., 2005, and the region of MLF1 contains a NES sequence and two putative NLS sequences. NPM-MLF1, recruited to the nucleolus, has the ability to override cellular senescence and to facilitate oncogenic transformation. It impairs the expression of p53 in response to genotoxic or oncogenic stress. The expression of NPM-MLF1 in MEFs facilitates an early escape from senescence and induces oncogenic transformation in collaboration with Ras (Yoneda-Kato and Kato, 2008) ( Figure 3 ).
Other facets of cop1
Protein is a variable molecule. Therefore, it will be abortive thinking that COP1 played only one type of role. According to Migliorini et al., 2011 COP1 plays role as a tumor suppressor through degrading c-JUN in genetically engineered mice. Although, there presents species-specific wiring variation between human and mice Migliorini et al.,2011. Besides COP1 implicated in fasting & hepatic glucose metabolism and fatty acid synthesis through degrading CREBregulated transcription coactivator-2 (CRTC2), forkhead box protein01 (FOXO1) and acetyl-CoA carboxylase (ACC) respectively Marine, 2012.
Conclusion
COP1 is a critical oncogene that plays pivotal role to grow cancer cells. COP1 can initiate cancer growth without p53’s mutation even without MDM2 gene amplification, although these two events consider as major cause of cancer. Therefore, COP1 demands great effort to elucidate all undiscovered pathways and partially discovered pathways. How COP1 mediates p53’s nuclear export demands elucidation. It arouses a question if there is any direct or indirect relationship between PTEN and COP1. COP1 down regulates 14-3- 3σ’s expression. It is to be clarified that COP1 overexpression causes 14-3-3σ’s expression lower or not. Another thing demands more specific result that when ATM is absent, whether ATR, DNA-PK and Jun kinases phosphorylate on COP1 at s387 and CSN3 or not. It should be made clearer how Ras signaling pathway links to COP1 pathways. Searching suitable therapeutics against COP1 is the laborious emerging field.
Abbreviations
Akt / PKB: Protein kinase B, AML: acute myeloid leukemia, AT: Ataxia telangiectasia, ATM: Ataxia telangiectasia mutated, ATR: ataxia telangiectasia and Rad3- related protein, ARF: Alternate Reading Frame, Arg: Arginine, CDC2/cyclinB: Cell division control 2, COP1: Constitutive Photomorphogenic Protein 1, CRM1: Chromosome Region Maintenance1, CSN3: COP9 signalosome complex subunit 3, CSN6: COP9 signalosome complex subunit 6, DNA-PK: DNAdependent protein kinase, Gy: Geiger, Glu: Glutamine, HCC: hepatocellular carcinomas, 14-3-3σ: Human mammary epithelium marker 1, HY5: Hypocotyl 5, IR: ionizing radiation, MDM2: Mouse Double Minute-2, MEF: mouse embryo fibroblast, MLF1: Myeloid leukemia factor 1, mTOR: mammalian target of rapamycin, NLS: Nuclear localization sequence, NES: Nuclear export sequence, P: proline, PIK: phosphatidylinositol phosphate kinase, PI3-K: phosphatidylinositol 3- phosphate kinase, Pirh2: p53-induced protein with a RING-h2 domain, NPM: nucleophosmin, NPMMLF1: nucleophosmin – Myeloid leukemia factor 1 fusion protein, PTEN: phosphatase and tensin homologue, PI3K: phosphatidylinositide 3-OH kinase, RFWD2: Ring finger and WD repeat domain 2, SiRNA: small interfering RNA, SFN: Stratifin, Ser: Serine, T: Threonine, TORC2: cAMP responsive coactivator, TP53: Tumor suppressor gene.
References
-
C.W.
Anderson,
T.H.
Carter.
The DNA-activated protein kinase-DNA-PK. In Molecular Analysis of DNA Rearrangements in the Immune System. Springer.
1996;
:
91-111
.
-
E.
Bianchi,
S.
Denti,
R.
Catena,
G.
Rossetti,
S.
Polo,
S.
Gasparian,
S.
Putignano,
L.
Rogge,
R.
Pardi.
Characterization of human constitutive photomorphogenesis protein 1, a RING finger ubiquitin ligase that interacts with Jun transcription factors and modulates their transcriptional activity. Journal of Biological Chemistry.
2003;
278
:
19682-19690
.
-
T.A.
Chan,
H.
Hermeking,
C.
Lengauer,
K.W.
Kinzler,
B.
Vogelstein.
14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature.
1999;
401
:
616-620
.
-
B.
Chen,
R.
Zhao,
C.-H.
Su,
M.
Linan,
C.
Tseng,
L.
Phan,
L.
Fang,
H.-Y.
Yang,
H.
Yang,
W.
Wang.
CDK inhibitor p57 (Kip2) is negatively regulated by COP9 signalosome subunit 6. Cell Cycle.
2012;
11
:
4633-4641
.
-
D.
Dornan,
S.
Bheddah,
K.
Newton,
W.
Ince,
G.D.
Frantz,
P.
Dowd,
H.
Koeppen,
V.M.
Dixit,
D.M.
French.
COP1, the negative regulator of p53, is overexpressed in breast and ovarian adenocarcinomas. Cancer research.
2004a;
64
:
7226-7230
.
-
D.
Dornan,
H.
Shimizu,
A.
Mah,
T.
Dudhela,
M.
Eby,
K.
O'Rourke,
S.
Seshagiri,
V.M.
Dixit.
ATM engages autodegradation of the E3 ubiquitin ligase COP1 after DNA damage. Science.
2006;
313
:
1122-1126
.
-
D.
Dornan,
I.
Wertz,
H.
Shimizu,
D.
Arnott,
G.D.
Frantz,
P.
Dowd,
K.
O'Rourke,
H.
Koeppen,
V.M.
Dixit.
The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature.
2004b;
429
:
86-92
.
-
J.
Feng,
R.
Tamaskovic,
Z.
Yang,
D.P.
Brazil,
A.
Merlo,
D.
Hess,
B.A.
Hemmings.
Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Aktdependent phosphorylation. Journal of Biological Chemistry.
2004;
279
:
35510-35517
.
-
A.T.
Ferguson,
E.
Evron,
C.B.
Umbricht,
T.K.
Pandita,
T.A.
Chan,
H.
Hermeking,
J.R.
Marks,
A.R.
Lambers,
P.A.
Futreal,
M.R.
Stampfer.
High frequency of hypermethylation at the 14-3-3 σ locus leads to gene silencing in breast cancer. Proceedings of the National Academy of Sciences.
2000;
97
:
6049-6054
.
-
D.J.
Freeman,
A.G.
Li,
G.
Wei,
H.-H.
Li,
N.
Kertesz,
R.
Lesche,
A.D.
Whale,
H.
Martinez-Diaz,
N.
Rozengurt,
R.D.
Cardiff.
PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent andindependent mechanisms. Cancer cell.
2003;
3
:
117-130
.
-
M.
Fukuda,
S.
Asano,
T.
Nakamura,
M.
Adachi,
M.
Yoshida,
M.
Yanagida,
E.
Nishida.
CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature.
1997;
390
:
308-311
.
-
A.
Fukumoto,
K.
Tomoda,
M.
Kubota,
J.Y.
Kato,
N.
Yoneda- Kato.
Small Jab1-containing subcomplex is regulated in an anchorage- and cell cycle-dependent manner, which is abrogated by ras transformation. FEBS letters.
2005;
579
:
1047-1054
.
-
C.S.
Hardtke,
K.
Gohda,
M.T.
Osterlund,
T.
Oyama,
K.
Okada,
X.W.
Deng.
HY5 stability and activity in Arabidopsis is regulated by phosphorylation in its COP1 binding domain. The EMBO journal.
2000;
19
:
4997-5006
.
-
H.
Hermeking,
C.
Lengauer,
K.
Polyak,
T.C.
He,
L.
Zhang,
S.
Thiagalingam,
K.W.
Kinzler,
B.
Vogelstein.
14-3- 3 sigma is a p53-regulated inhibitor of G2/M progression. Molecular cell.
1997;
1
:
3-11
.
-
N.
Iwata,
H.
Yamamoto,
S.
Sasaki,
F.
Itoh,
H.
Suzuki,
T.
Kikuchi,
H.
Kaneto,
S.
Iku,
I.
Ozeki,
Y.
Karino.
Frequent hypermethylation of CpG islands and loss of expression of the 14-3-3 sigma gene in human hepatocellular carcinoma. Oncogene.
2000;
19
:
5298-5302
.
-
C.T.
Keith,
S.L.
Schreiber.
PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. SCIENCE-NEW YORK THEN WASHINGTON-.
1995;
:
50-50
.
-
D.P.
Lane.
Cancer. p53, guardian of the genome. Nature.
1992;
358
:
15-16
.
-
C.
Laronga,
H.-Y.
Yang,
C.
Neal,
M.-H.
Lee.
Association of the cyclin-dependent kinases and 14-3-3 sigma negatively regulates cell cycle progression. Journal of Biological Chemistry.
2000;
275
:
23106-23112
.
-
M.F.
Lavin,
S.
Kozlov.
ATM activation and DNA damage response. Cell cycle.
2007;
6
:
931-942
.
-
M.-H.
Lee,
R.
Zhao,
L.
Phan,
S.-C.J.
Yeung.
Roles of COP9 signalosome in cancer. Cell cycle.
2011;
10
:
305-7
.
-
Y.-H.
Lee,
J.B.
Andersen,
H.-T.
Song,
A.D.
Judge,
D.
Seo,
T.
Ishikawa,
J.U.
Marquardt,
M.
Kitade,
M.E.
Durkin,
C.
Raggi.
Definition of ubiquitination modulator COP1 as a novel therapeutic target in human hepatocellular carcinoma. Cancer research.
2010;
70
:
8264-8269
.
-
S.
Lees-Miller,
K.
Sakaguchi,
S.
Ullrich,
E.
Appella,
C.
Anderson.
Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Molecular and cellular biology.
1992;
12
:
5041-5049
.
-
Y.F.
Li,
D.D.
Wang,
B.W.
Zhao,
W.
Wang,
C.Y.
Huang,
Y.M.
Chen,
Y.
Zheng,
R.P.
Keshari,
J.C.
Xia,
Z.W.
Zhou.
High level of COP1 expression is associated with poor prognosis in primary gastric cancer. International journal of biological sciences.
2012;
8
:
1168-1177
.
-
X.
Lu.
Tied up in loops: positive and negative autoregulation of p53. Cold Spring Harbor perspectives in biology.
2010;
2
:
a000984
.
-
J.-C.
Marine.
Spotlight on the role of COP1 in tumorigenesis. Nature Reviews Cancer.
2012;
12
:
455-464
.
-
D.
Migliorini,
S.
Bogaerts,
D.
Defever,
R.
Vyas,
G.
Denecker,
E.
Radaelli,
A.
Zwolinska,
V.
Depaepe,
T.
Hochepied,
W.C.
Skarnes.
Cop1 constitutively regulates c-Jun protein stability and functions as a tumor suppressor in mice. The Journal of clinical investigation.
2011;
121
:
1329-1343
.
-
Y.
Pereg,
D.
Shkedy,
P.
de Graaf,
E.
Meulmeester,
M.
Edelson- Averbukh,
M.
Salek,
S.
Biton,
A.F.
Teunisse,
W.D.
Lehmann,
A.G.
Jochemsen.
Phosphorylation of Hdmx mediates its Hdm2-and ATM-dependent degradation in response to DNA damage. Proceedings of the National Academy of Sciences of the United States of America.
2005;
102
:
5056-5061
.
-
G.L.
Prasad,
E.M.
Valverius,
E.
McDuffie,
H.
Cooper.
Complementary DNA cloning of a novel epithelial cell marker protein, HME1, that may be down-regulated in neoplastic mammary cells. Cell growth and differentiation.
1992;
3
:
507-507
.
-
J.
Puc,
M.
Keniry,
H.S.
Li,
T.K.
Pandita,
A.D.
Choudhury,
L.
Memeo,
M.
Mansukhani,
V.V.
Murty,
Z.
Gaciong,
S.E.
Meek.
Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer cell.
2005;
7
:
193-204
.
-
J.D.
Siliciano,
C.E.
Canman,
Y.
Taya,
K.
Sakaguchi,
E.
Appella,
M.B.
Kastan.
DNA damage induces phosphorylation of the amino terminus of p53. Genes & development.
1997;
11
:
3471-3481
.
-
C.H.
Su,
R.
Zhao,
G.
Velazquez-Torres,
J.
Chen,
C.
Gully,
S.C.
Yeung,
M.H.
Lee.
Nuclear export regulation of COP1 by 14-3-3sigma in response to DNA damage. Molecular cancer.
2010;
9
:
24-3
.
-
R.S.
Tibbetts,
K.M.
Brumbaugh,
J.M.
Williams,
J.N.
Sarkaria,
W.A.
Cliby,
S.-Y.
Shieh,
Y.
Taya,
C.
Prives,
R.T.
Abraham.
A role for ATR in the DNA damage-induced phosphorylation of p53. Genes & development.
1999;
13
:
152-157
.
-
I.
Vivanco,
C.L.
Sawyers.
The phosphatidylinositol 3- kinase-AKT pathway in human cancer. Nature Reviews Cancer.
2002;
2
:
489-501
.
-
A.
Von Arnim,
X.-W.
Deng.
Ring finger motif of Arabidopsis thaliana COP1 defines a new class of zinc-binding domain. Journal of Biological Chemistry.
1993;
268
:
19626-19631
.
-
W.
Wang,
A.
Budhu,
M.
Forgues,
X.W.
Wang.
Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nature Cell Biology.
2005;
7
:
823-830
.
-
M.J.
Waterman,
E.S.
Stavridi,
J.L.
Waterman,
T.D.
Halazonetis.
ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nature genetics.
1998;
19
:
175-178
.
-
K.B.
Wee,
B.D.
Aguda.
Akt versus p53 in a network of oncogenes and tumor suppressor genes regulating cell survival and death. Biophysical journal.
2006;
91
:
857-865
.
-
E.W.
Wilker,
R.A.
Grant,
S.C.
Artim,
M.B.
Yaffe.
A structural basis for 14-3-3sigma functional specificity. The Journal of biological chemistry.
2005;
280
:
18891-18898
.
-
T.
Yamada,
K.
Christov,
A.
Shilkaitis,
L.
Bratescu,
A.
Green,
S.
Santini,
A.
Bizzarri,
S.
Cannistraro,
T.
Gupta,
C.
Beattie.
p28, A first in class peptide inhibitor of cop1 binding to p53. British journal of cancer.
2013;
108
:
2495-2504
.
-
H.
Yang,
Y.
Wen,
Y.
Lin,
L.
Pham,
C.
Su,
H.
Yang,
J.
Chen,
M.
Lee.
Roles for negative cell regulator 14-3-3σ in control of MDM2 activities. Oncogene.
2007;
26
:
7355-7362
.
-
H.
Yang,
Y.-Y.
Wen,
R.
Zhao,
Y.-L.
Lin,
K.
Fournier,
H.-Y.
Yang,
Y.
Qiu,
J.
Diaz,
C.
Laronga,
M.-H.
Lee.
DNA Damage-Induced Protein 14-3-3 σ Inhibits Protein Kinase B/Akt Activation and Suppresses Akt-Activated Cancer. Cancer research.
2006;
66
:
3096-3105
.
-
H.-Y.
Yang,
Y.-Y.
Wen,
C.-H.
Chen,
G.
Lozano,
M.-H.
Lee.
14-3-3σ positively regulates p53 and suppresses tumor growth. Molecular and cellular biology.
2003;
23
:
7096-7107
.
-
N.
Yoneda-Kato,
J.-y.
Kato.
Shuttling imbalance of MLF1 results in p53 instability and increases susceptibility to oncogenic transformation. Molecular and cellular biology.
2008;
28
:
422-434
.
-
N.
Yoneda-Kato,
A.T.
Look,
M.N.
Kirstein,
M.B.
Valentine,
S.C.
Raimondi,
K.J.
Cohen,
A.J.
Carroll,
S.W.
Morris.
The t (3; 5)(q25. 1; q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPMMLF1. Oncogene.
1996;
12
:
265-275
.
-
N.
Yoneda-Kato,
K.
Tomoda,
M.
Umehara,
Y.
Arata,
J.y.
Kato.
Myeloid leukemia factor 1 regulates p53 by suppressing COP1 via COP9 signalosome subunit 3. The EMBO journal.
2005;
24
:
1739-1749
.
-
B.P.
Zhou,
Y.
Liao,
W.
Xia,
Y.
Zou,
B.
Spohn,
M.-C.
Hung.
HER-2/neu induces p53 ubiquitination via Aktmediated MDM2 phosphorylation. Nature cell biology.
2001;
3
:
973-982
.
-
M.
Rabbani,
S.
Hossain,
K.
Islam,
S.
& Uddin.
Constitutive Photomorphogensis Protein1 (COP1) mediated p53 pathway and its oncogenic role. Biomedical Research And.
2014;
Therapy
:
1(5), 142-151
.
Comments
Downloads
Article Details
Volume & Issue : Vol 1 No 05 (2014)
Page No.: 142-151
Published on: 2014-12-09
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 5534 times
- Download PDF downloaded - 1954 times
- View Article downloaded - 10 times

{kind=link}
{kind=link}
{kind=link}
{kind=link}