

Neuroimmune Dysregulation as a Public Health Axis in Neurodegeneration: Decoding Immune Checkpoints, Aging, and Biomarkers in Preclinical Alzheimer’s Disease
- Shri Venkateshwara University, India
Abstract
Alzheimer’s Disease (AD), the leading cause of dementia globally, is increasingly recognized as a multifactorial disorder in which neurodegeneration intersects with systemic immune aging. While classical models emphasize amyloid-β and tau accumulation, accumulating evidence suggests that neuroimmune dysregulation may play a critical role during the earliest, preclinical stages of disease development. This review synthesizes emerging literature on how immune aging, immune checkpoint signaling, and biomarker discovery converge to shape early AD pathophysiology. Age-associated immune remodeling including T-cell exhaustion, impaired antigen presentation, and altered innate immune responses can disrupt central nervous system immune surveillance. In particular, dysregulated signaling through checkpoint molecules such as Programmed Cell Death Protein 1 and Cytotoxic T-Lymphocyte Associated Protein 4 may influence microglial activation states, peripheral immune infiltration, and neuroinflammatory cascades that precede overt neurodegeneration. Advances in neuroimmunology and biomarker technologies including cerebrospinal fluid immune profiling, peripheral immunophenotyping, and molecular neuroimaging are beginning to reveal measurable neuroimmune signatures associated with early disease states. However, important challenges remain regarding biomarker specificity, translational feasibility, and implementation across diverse healthcare systems. By integrating insights from immunology, neuroscience, and public health research, this review proposes that neuroimmune dysfunction represents a trackable component of early AD pathogenesis. Understanding how immune aging interacts with neurodegenerative pathways may help inform future strategies for early risk stratification and preventative intervention.
INTRODUCTION
Alzheimer’s Disease (AD) is a progressive neurodegenerative disorder and the leading cause of dementia worldwide, representing one of the most pressing neurological and public health challenges of the twenty-first century. Recent global estimates indicate that over 55 million individuals are currently living with dementia, with AD accounting for approximately 60-70% of cases, a burden projected to rise substantially with population aging and increased life expectancy1,2,3. Despite decades of intensive research, therapeutic strategies targeting the classical pathological hallmarks of AD amyloid-β plaques and tau neurofibrillary tangles have demonstrated only modest or inconsistent clinical benefits, particularly when administered after the onset of symptoms4,5,6. These limitations have prompted a paradigm shift toward understanding AD as a multifactorial disorder involving complex interactions between neurodegenerative and systemic processes. In this evolving framework, neuroimmune dysregulation has emerged as a critical contributor to early disease pathogenesis7,8,9. Increasing evidence suggests that neurodegeneration unfolds within a broader context of age-associated immune remodeling, encompassing both central and peripheral immune compartments. Alterations in innate immune cells, particularly microglia, along with dysregulated adaptive immune responses, contribute to the establishment of a chronic neuroinflammatory milieu that may precede overt neuronal damage10,11,12. Peripheral immune components, including T lymphocytes and monocyte-derived macrophages, further participate in this process, reflecting dynamic bidirectional communication along the neuroimmune axis13,14.
Within this context, immune checkpoint signaling pathways have gained increasing attention for their dual roles in maintaining immune homeostasis and modulating neuroinflammatory responses. Molecules such as programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) are essential for preserving immune tolerance and preventing excessive immune activation; however, their age-associated upregulation may also contribute to impaired immune surveillance and diminished capacity for tissue repair in the central nervous system15,16,17. Experimental and translational studies suggest that modulation of these pathways can influence microglial activation states, peripheral immune cell infiltration, and amyloid clearance mechanisms, although the clinical applicability of such approaches remains under active investigation18,19. Parallel advances in biomarker research are transforming the study of early AD. Technologies such as cerebrospinal fluid immune profiling, peripheral immunophenotyping, and molecular neuroimaging targeting neuroinflammation are beginning to reveal measurable neuroimmune signatures associated with preclinical disease stages10,11. Emerging analytical platforms, including high-resolution spectroscopic techniques, further offer the potential to detect metabolic and molecular signatures linked to immune aging and cellular stress responses, although these approaches remain largely investigational.
Importantly, the intersection of aging biology and immune regulation provides a broader mechanistic context for these observations. Immunosenescence, defined as the progressive decline in immune competence with aging, is characterized by features such as T-cell exhaustion, altered cytokine production, and epigenetic remodeling of immune gene expression12,13. Recent evidence indicates that age-associated epigenetic alterations including DNA methylation changes at immune regulatory loci such as PDCD1 and histone modifications influencing microglial genes like TREM2 may contribute to sustained immune dysfunction and chronic neuroinflammation in the aging brain14,15,16. These findings support the concept that neuroimmune dysregulation represents a fundamental component of AD susceptibility rather than merely a secondary response to neuronal injury. From a public health perspective, recognizing the contribution of immune dysfunction to AD pathogenesis underscores the importance of early detection and risk stratification strategies. However, translating immune-based biomarkers and immunomodulatory interventions into routine clinical practice remains challenging. Limitations in biomarker specificity, lack of standardized assay platforms, and variability across patient populations present significant barriers to clinical implementation17,18,19. These challenges are particularly pronounced in low- and middle-income countries, where limited healthcare infrastructure, economic constraints, and a high burden of infectious diseases may further complicate large-scale deployment of advanced diagnostic technologies.
In this context, the present review synthesizes current evidence on neuroimmune mechanisms in the early stages of AD, focusing on four interconnected themes: (i) the molecular and cellular roles of immune checkpoint signaling in neurodegenerative processes; (ii) the influence of immune aging and immunosenescence on AD susceptibility; (iii) emerging biomarkers reflecting neuroimmune dysfunction; and (iv) the translational opportunities and limitations associated with immune-targeted therapeutic strategies. By integrating insights from immunology, neuroscience, and translational research, this review aims to clarify how immune regulatory pathways contribute to the earliest phases of AD and to identify critical knowledge gaps that must be addressed to advance preventative and therapeutic approaches.
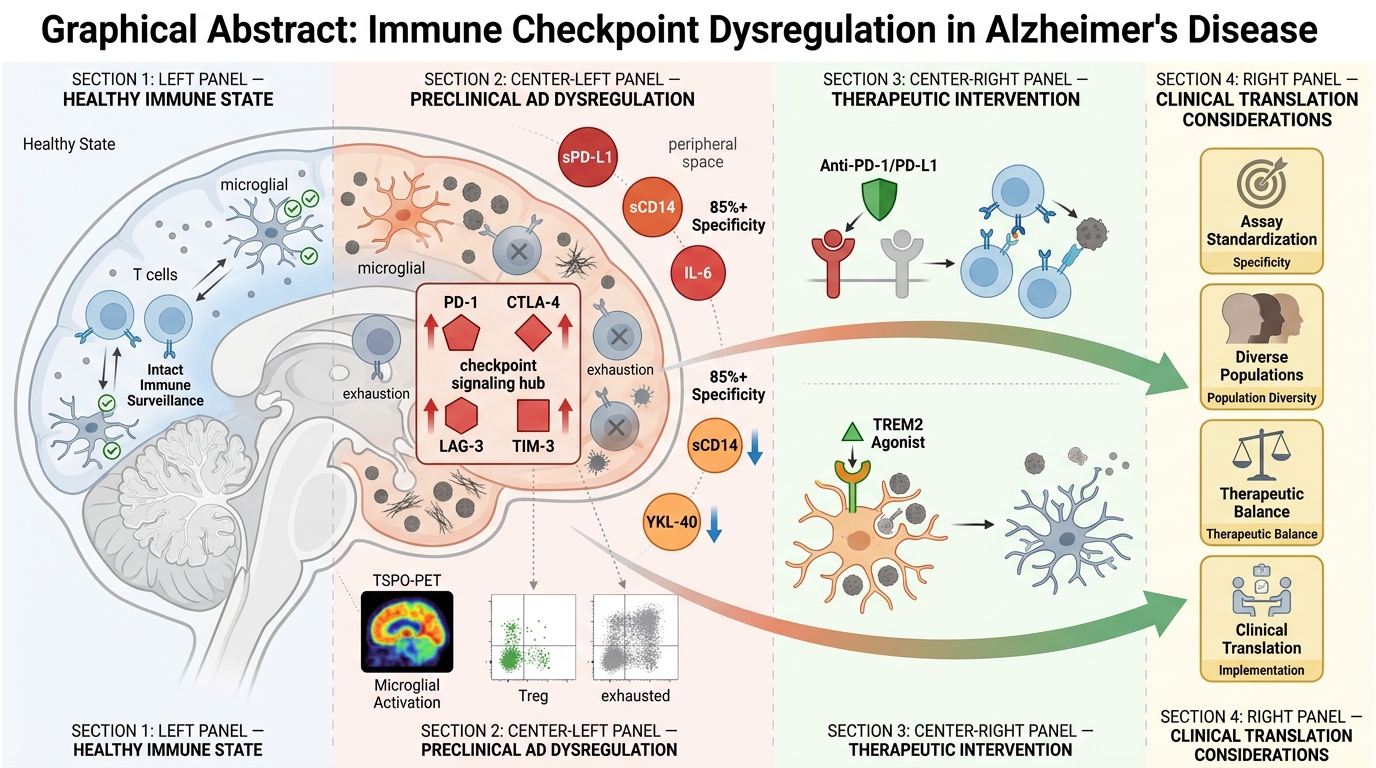
By decoding the interface of immune aging and neurodegeneration, this work aims to provide a roadmap for immune-informed prevention strategies in AD bridging basic immunology, biomarker science, and translational neurology to redefine therapeutic paradigms. Figure 1 schematically illustrates the progressive neuroimmune remodeling underlying Alzheimer’s Disease, delineating how escalating microglial activation, immune checkpoint–mediated immunosuppression, and peripheral immune biomarker signatures converge with advanced neuroimaging and multi-omic platforms to enable early diagnosis, risk stratification, and scalable public health implementation across diverse aging populations.
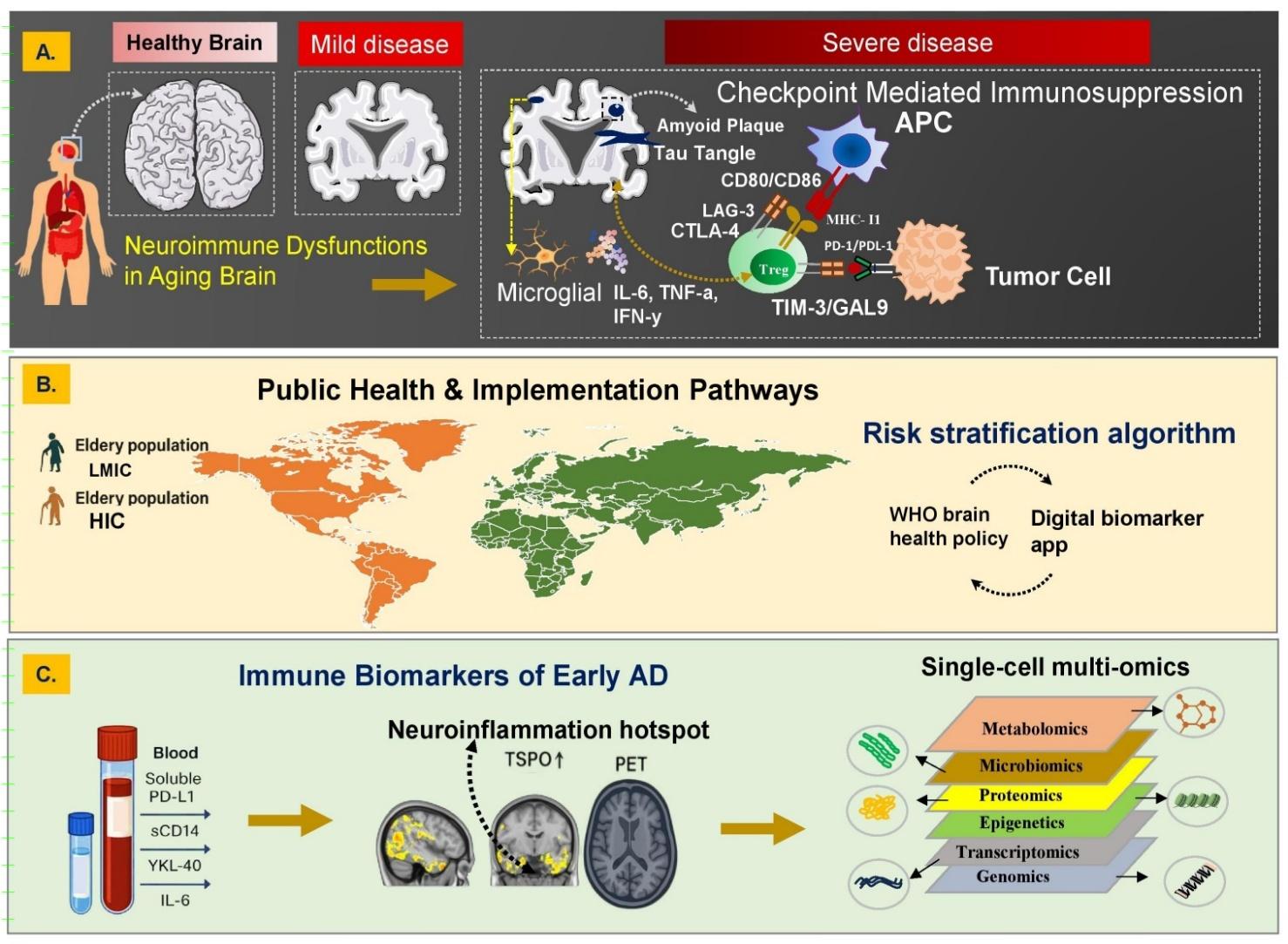
Neuroimmune Continuum of Alzheimer’s Disease Progression: From Immune Dysregulation to Translational Precision Diagnostics and Global Health Integration. (A) Age-associated neuroimmune dysfunction characterized by microglial activation, pro-inflammatory cytokines (IL-6, TNF-α, IFN-γ), and immune checkpoint signaling (PD-1/PD-L1, CTLA-4, LAG-3, TIM-3/GAL-9) drives impaired immune surveillance, promoting amyloid and tau pathology with progressive neurodegeneration. (B) Population-level implementation framework integrating neuroimmune risk stratification with digital biomarkers and global brain health policies to enable scalable, equitable early detection and prevention across LMICs and HICs. (C) Blood-based immune biomarkers (e.g., soluble PD-L1, sCD14, YKL-40, IL-6) linked to TSPO-PET neuroinflammation, integrated with multi-omic layers to support early diagnosis, mechanistic stratification, and precision neuroimmune profiling. Abbreviations: AD: Alzheimer’s Disease; IL-6: Interleukin-6; TNF-α: Tumor necrosis factor-alpha; IFN-γ: Interferon-gamma; PD-1: Programmed cell death protein 1; PD-L1: Programmed death-ligand 1; CTLA-4: Cytotoxic T-lymphocyte–associated protein 4; LAG-3: Lymphocyte activation gene-3; TIM-3: T-cell immunoglobulin and mucin-domain containing-3; GAL-9: Galectin-9; LMICs: Low- and middle-income countries; HICs: High-income countries; sCD14: Soluble CD14; YKL-40: Chitinase-3-like protein 1; TSPO: Translocator protein (18 kDa); PET: Positron emission tomography.
Literature Search Strategy and Study Selection
To ensure transparency and minimize selection bias, this review followed a structured literature identification and synthesis strategy aligned with recommendations from the EQUATOR Network and the methodological principles outlined in the SANRA (Scale for the Assessment of Narrative Review Articles). The present study is designed as a narrative integrative review and does not adhere to formal systematic review or scoping review reporting frameworks.
Database Search Strategy
A comprehensive literature search was conducted across major biomedical databases including PubMed/MEDLINE, Scopus, and Web of Science to identify peer-reviewed articles relevant to neuroimmune mechanisms in Alzheimer’s Disease. The search primarily focused on publications from January 2000 to March 2026, with particular emphasis on studies published in the past decade reflecting advances in neuroimmunology, biomarker development, and translational therapeutics.
Search queries combined controlled vocabulary and keyword-based terms related to neuroimmune regulation and immune checkpoints. Representative search strings included combinations of:
-
“Alzheimer’s Disease” AND “neuroinflammation”
-
“Immune checkpoints” OR Programmed Cell Death Protein 1 OR Cytotoxic T-Lymphocyte Associated Protein 4
-
“Microglial activation”
-
“Immunosenescence”
-
“T-cell exhaustion”
-
“Neuroimmune biomarkers”
-
“Preclinical Alzheimer’s Disease”
Reference lists of key review articles and landmark experimental studies were additionally screened to identify relevant publications not captured during the initial database search.
Inclusion and Exclusion Criteria
Studies were included if they met the following criteria:
-
Peer-reviewed research articles, clinical studies, or authoritative reviews addressing neuroimmune mechanisms relevant to AD pathogenesis.
-
Studies investigating immune checkpoint signaling, microglial activation, Immunosenescence, or immune biomarkers in the context of neurodegeneration.
-
Preclinical experimental models, human observational studies, clinical biomarker investigations, or translational immunotherapy research related to AD.
Articles were excluded if they:
-
focused exclusively on non-AD neurodegenerative disorders without mechanistic relevance to AD;
-
lacked primary mechanistic or clinical insight into neuroimmune pathways;
-
were non-peer-reviewed sources, editorials, or conference abstracts without sufficient methodological detail.
Evidence Evaluation and Risk of Bias Considerations
Given the heterogeneity of available evidence including preclinical mechanistic studies, observational biomarker investigations, and early-phase therapeutic trials formal quantitative risk-of-bias scoring was not applied. Instead, studies were critically evaluated based on methodological robustness, sample size, reproducibility across independent cohorts, and translational relevance to human disease. Priority was given to large cohort biomarker studies, well-characterized experimental models, and peer-reviewed clinical investigations. Conflicting findings within the literature were considered and discussed where relevant to highlight areas of ongoing debate, particularly in the context of immune checkpoint modulation and neuroinflammatory signaling in AD.
Data Synthesis
The selected literature was synthesized thematically to examine three interconnected dimensions of neuroimmune dysfunction in AD:
-
age-associated immune remodeling and Immunosenescence,
-
dysregulation of immune checkpoint pathways in the central nervous system, and
-
emerging neuroimmune biomarkers with potential relevance for early disease detection and risk stratification.
This integrative approach enables the contextualization of molecular mechanisms alongside translational implications for biomarker development and future immunomodulatory strategies.
THE GLOBAL BURDEN OF PRECLINICAL ALZHEIMER’S DISEASE
Alzheimer’s Disease (AD) has traditionally been conceptualized as the clinical endpoint of progressive neurodegeneration, characterized by memory impairment, cognitive decline, and functional deterioration. However, converging evidence from longitudinal cohorts and biomarker-driven studies indicates that AD pathogenesis begins decades prior to symptom onset, within a prolonged preclinical phase marked by silent yet progressive neurobiological alterations20. These changes include the gradual accumulation of amyloid-β (Aβ), early tau hyperphosphorylation, mitochondrial dysfunction, synaptic impairment, and critically, neuroimmune remodeling21,22. Accordingly, the true global burden of AD extends far beyond clinically manifest disease and encompasses a substantial, yet largely unrecognized, reservoir of preclinical pathology.
Advanced age is the strongest and most well-established non-modifiable risk factor for Alzheimer’s Disease (AD) and its preclinical forms, as consistently demonstrated across large-scale epidemiological and longitudinal studies Lancet Commission on Dementia Prevention, Intervention, and Care23. Parallel to this, global demographic shifts are accelerating disease burden, with the population aged ≥60 years projected to increase from approximately 1 billion in 2020 to 2.1 billion by 2050, thereby amplifying the prevalence of preclinical AD on an unprecedented scale24. Cognitive aging, once considered a benign and inevitable process, is now increasingly understood as a continuum underpinned by progressive neuropathological changes. Landmark epidemiological studies, including the Mayo Clinic Study of Aging and the Framingham Heart Study, have demonstrated that approximately 30-40% of cognitively normal older adults harbor biomarker evidence of Aβ deposition or tau pathology, fulfilling criteria for preclinical AD25,26.
Importantly, this silent neuropathological burden is not confined to high-income countries (HICs). Low- and middle-income countries (LMICs), which are undergoing rapid demographic and epidemiological transitions, are projected to bear a disproportionate share of the global dementia burden. By 2050, nearly 70-71% of individuals with dementia are expected to reside in LMICs, where limitations in diagnostic infrastructure, specialist access, and disease awareness persist World Health Organization27. The convergence of increased life expectancy, suboptimal cardiovascular risk management, metabolic comorbidities, and chronic systemic inflammation in these regions accelerates the progression from preclinical pathology to overt neurodegeneration28,29. Additional modifiers including environmental exposures, lower educational attainment, and diminished cognitive reserve further exacerbate susceptibility to disease onset and progression30.
Despite these advances, early diagnosis of Alzheimer’s Disease remains a critical unmet need in global public health. Current diagnostic paradigms are largely predicated on the onset of clinical symptoms, relying on neuropsychological assessment and caregiver-reported cognitive decline, thereby failing to identify individuals in the asymptomatic but biologically active stages of disease31. Although biomarker-based approaches, including cerebrospinal fluid (CSF) assays and amyloid/tau positron emission tomography (PET), offer high sensitivity for early detection, their widespread implementation is constrained by cost, infrastructure requirements, and limited technical expertise, particularly in resource-limited settings32. This diagnostic gap perpetuates global inequities and delays the initiation of preventive or disease-modifying strategies during the earliest and most therapeutically tractable phase of AD.
In this context, current global health strategies and dementia care frameworks remain predominantly oriented toward symptomatic diagnosis and management, with limited integration of structured screening pathways for individuals at risk of preclinical Alzheimer’s Disease based on age, genetic susceptibility (e.g., APOE ε4), or emerging metabolic and immune biomarkers. This reflects a broader implementation gap between advancing biomarker science and real-world healthcare policy adoption, particularly in low- and middle-income countries33,34. An underappreciated yet transformative factor in this landscape is the emerging recognition of neuroimmune dysregulation as a sentinel biomarker and driver of preclinical AD. Studies from both human postmortem brains and longitudinal cohort data (e.g., the Religious Orders Study and the Alzheimer’s Disease Neuroimaging Initiative) have shown that elevations in peripheral pro-inflammatory cytokines, T cell exhaustion markers, and microglial activation occur prior to measurable cognitive decline35,36,37. These immune features not only correlate with amyloid and tau deposition but may also contribute causally to disease initiation by disrupting homeostatic clearance, impairing synaptic pruning, and accelerating neurovascular dysfunction38.
In LMICs, where access to PET imaging and lumbar punctures is limited, the integration of immune biomarkers and blood-based spectroscopic tools offers a more scalable and equitable approach to early detection. Raman spectroscopy, FTIR, and NMR-based platforms can detect epigenetic and metabolic signatures of immune aging and T cell dysfunction, offering a non-invasive, low-cost alternative to traditional biomarker pipelines39,40. These technologies are particularly relevant in public health surveillance contexts, where high-throughput, rapid screening could facilitate early risk stratification and longitudinal tracking. Socioeconomic inequities also compound the challenge of addressing preclinical AD globally. In many LMICs, individuals at risk of cognitive decline have limited access to preventive healthcare, insurance coverage, or cognitive rehabilitation services. Furthermore, gendered patterns of health-seeking behaviour, education, and caregiving mean that women who constitute two-thirds of AD cases are particularly vulnerable to delayed diagnosis and intervention41. These disparities are compounded by structural underinvestment in mental health infrastructure, fragmented elder care systems, and poor integration of geriatric and neurological services42. Despite the scale of the problem, opportunities for intervention abound. Immune-targeted therapies, including checkpoint inhibitors and immune rejuvenation agents, have shown early promise in preclinical AD models and small human trials, suggesting that targeting neuroimmune failure may delay the onset or progression of disease43,44. Furthermore, population-level interventions such as cardiovascular risk control, diet modification, cognitive stimulation, and anti-inflammatory lifestyle measures may reduce the cumulative burden of neuroimmune stressors, especially if initiated during midlife45. Framing preclinical AD as a modifiable and immunologically active stage encourages a shift from reactive to preventive paradigms in both policy and practice.
Ultimately, addressing the global burden of preclinical AD requires an integrated, multidisciplinary strategy that bridges clinical neuroscience, public health policy, molecular immunology, and health equity frameworks. This includes: (i) redefining diagnostic criteria to incorporate immune and metabolic risk profiles; (ii) promoting biomarker development tailored for low-resource settings; (iii) embedding cognitive and immune health into primary care and aging programs; and (iv) fostering global research collaborations focused on diverse populations. A failure to act now will result not only in escalating dementia prevalence but also in deepening social and health inequities across generations. To contextualize the multidimensional drivers underlying the global burden of preclinical Alzheimer’s Disease, Table 1 synthesizes key epidemiological, demographic, and healthcare-system determinants including population aging, silent disease prevalence, socioeconomic disparities, and health system capacity highlighting how these interconnected factors collectively shape disease vulnerability and diagnostic inequities across diverse global settings.
Global Determinants Influencing the Burden of Preclinical Alzheimer’s Disease
| Dimension | Key Determinants | Evidence From Literature | Public Health Implication |
|---|---|---|---|
| Population Aging | Increasing life expectancy and demographic transition | Aging is the strongest risk factor for AD and related dementias, with prevalence rising sharply after age 65 | Rapidly aging populations are expected to drive substantial increases in AD incidence globally |
| Silent or Preclinical Disease Prevalence | Long asymptomatic phase with progressive amyloid and tau accumulation | Neuropathological and biomarker studies show that pathological changes can begin decades before clinical symptoms | Early detection strategies targeting preclinical stages are critical for prevention |
| Socioeconomic and Healthcare Disparities | Limited access to diagnostic infrastructure, specialist care, and biomarker testing | Lower diagnostic rates and delayed clinical recognition are common in resource-limited settings | Health system strengthening and improved diagnostic accessibility are required |
| Public Health System Capacity | Workforce shortages, limited geriatric and neurological care resources | Many healthcare systems lack adequate dementia screening and long-term care programs | Integrating dementia risk monitoring into primary healthcare systems may improve early identification |
IMMUNE CHECKPOINT SIGNALING IN THE AGING BRAIN
The central nervous system (CNS) has long been regarded as an immune-privileged site; however, this paradigm has evolved to accommodate the dynamic and tightly regulated role of immune surveillance in maintaining neural homeostasis. Among the most critical regulators of CNS immune balance are immune checkpoint molecules, which fine-tune the activation thresholds of immune cells to prevent excessive inflammation or autoimmunity. In the context of aging and neurodegeneration, these regulatory pathways become increasingly dysregulated, contributing to the pathogenesis of Alzheimer’s Disease (AD) even before clinical symptoms arise. Immune checkpoint receptors such as programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte–associated protein 4 (CTLA-4), lymphocyte activation gene 3 (LAG-3), and T cell immunoglobulin and mucin-domain containing-3 (TIM-3) play central roles in modulating the activity of T lymphocytes, microglia, macrophages, and astrocytes within the aging brain. These molecules, typically upregulated during chronic immune stimulation, serve as co-inhibitory receptors that limit excessive immune activation. In the context of chronic neurodegenerative stress such as the presence of amyloid-β (Aβ) plaques or hyperphosphorylated tau checkpoint expression is often pathologically elevated, impairing immunosurveillance and fostering a permissive environment for neurodegeneration (Table 2).
Microglia, the resident innate immune cells of the CNS, express several immune checkpoint ligands including PD-L1 and galectin-9 (the ligand for TIM-3), especially under conditions of chronic inflammation and aging. In early AD, microglia undergo a functional transformation toward a disease-associated microglial (DAM) phenotype, characterized by impaired phagocytosis, altered metabolic states, and increased expression of immune checkpoint molecules46. This altered state diminishes their capacity to clear Aβ and debris, while simultaneously promoting chronic neuroinflammation via cytokine release. Similarly, astrocytes, once considered passive support cells, express checkpoint ligands such as B7 family members and also contribute to immune synapse formation and neuronal injury modulation47. In neuroinflammatory settings, astrocytes engage in cross-talk with infiltrating lymphocytes, further amplifying checkpoint-driven immunosuppression and reactive gliosis. Infiltrating peripheral T lymphocytes, especially CD8 and CD4 T cells, have been increasingly recognized as key players in CNS immune surveillance and AD pathology. With aging and chronic exposure to Aβ, these cells undergo functional exhaustion characterized by sustained expression of PD-1, LAG-3, and TIM-3, and reduced production of effector cytokines such as IL-2 and IFN-γ48. This immunosenescent phenotype limits the ability of T cells to mount effective anti-amyloid or neuroprotective responses, and may also impair clearance of viral reactivations such as latent herpesviruses factors implicated in AD progression. Moreover, CTLA-4 signaling in T regulatory cells (Tregs) becomes upregulated in aged individuals, potentially skewing the balance toward excessive immune tolerance in the brain parenchyma49.
Emerging evidence from human autopsy studies, cerebrospinal fluid (CSF) analyses, and single-cell transcriptomic profiling supports the dysregulation of immune checkpoints in early and preclinical AD. For instance, single-cell RNA sequencing of brain-infiltrating lymphocytes in individuals with mild cognitive impairment (MCI) has revealed elevated expression of PD-1, LAG-3, and TIM-3 transcripts in both CD8 and CD4 populations50. In parallel, microglial populations in preclinical AD brains exhibit upregulated PD-L1 and altered NF-κB signaling, suggesting a compensatory or maladaptive checkpoint engagement in response to early neuropathological triggers51. CSF samples from biomarker-positive but cognitively normal individuals show elevated levels of soluble PD-L1 and TIM-3 ligands, indicating systemic checkpoint activation even before clinical symptoms emerge52.
Checkpoint dysregulation does not occur in isolation; rather, it intersects intricately with the pathological hallmarks of AD amyloid-β, tau, and neuronal loss. PD-1-expressing T cells accumulate near amyloid plaques in both mouse models and human postmortem brains, yet appear functionally inert, unable to mount cytotoxic responses or promote clearance53. In murine models, blockade of PD-1 or CTLA-4 restores T cell effector function, enhances microglial phagocytosis of Aβ, and reduces plaque burden, suggesting a therapeutic window for immune reinvigoration54. Tau pathology, likewise, is exacerbated by chronic checkpoint-mediated immune paralysis. Recent data suggest that immune checkpoint activation in T cells and glial cells may impair tau clearance and potentiate the spread of tau aggregates via extracellular vesicles or compromised glymphatic flow52. In addition to promoting protein aggregation, checkpoint signaling may directly impact neuronal viability through glial-mediated mechanisms. Astrocytes and microglia, under the influence of checkpoint ligands, can adopt neurotoxic phenotypes that release glutamate, ROS, and pro-apoptotic mediators. PD-L1 expression in astrocytes has been shown to suppress synaptic remodeling and contribute to synapse loss in aging models, while CTLA-4 Tregs may limit the recruitment of protective immune cells to sites of neural injury55. These interactions reflect a broader landscape in which chronic immune checkpoint engagement fosters a milieu of immune exhaustion, failed clearance, and synaptic vulnerability. Collectively, these findings highlight that immune checkpoints in the aging brain are not simply inert markers of immune modulation but active participants in the pathophysiology of AD. The aberrant upregulation of PD-1, CTLA-4, TIM-3, and LAG-3 across immune and glial populations reflects a systemic breakdown in immune surveillance that precedes and potentially drives neurodegeneration. Targeting these pathways, either through localized immune checkpoint blockade, metabolic reprogramming, or combined epigenetic modulation, represents a promising frontier in the prevention of symptomatic AD. Understanding the temporal and spatial dynamics of checkpoint signaling particularly in the preclinical phase may offer critical insight into who is most at risk, when to intervene, and how best to restore neuroimmune equilibrium.
Translational Integration of Immune Checkpoint Biomarkers into Scalable Screening Frameworks in LMICs
To bridge the mechanistic insights of immune checkpoint dysregulation with real-world applicability, it is essential to contextualize how these molecular signatures can be translated into scalable diagnostic frameworks, particularly within low- and middle-income country (LMIC) settings. Emerging evidence suggests that peripheral immune alterations associated with early Alzheimer’s Disease including dysregulated expression of immune checkpoint receptors such as PD-1, LAG-3, and TIM-3 on circulating T cells, along with shifts in cytokine profiles (e.g., IL-6, TNF-α, IFN-γ) can serve as accessible and quantifiable biomarkers for preclinical disease detection50,52. These signatures can be integrated into multiplex blood-based biomarker panels, which are increasingly compatible with low-cost, high-throughput assay platforms such as ELISA-based arrays and point-of-care immunodiagnostic devices. In LMIC healthcare systems, where advanced neuroimaging and cerebrospinal fluid diagnostics remain limited, such minimally invasive and scalable approaches offer a pragmatic pathway for early risk stratification46,51. Coupling immune biomarker panels with digital health infrastructures including mobile health platforms and AI-assisted risk prediction algorithms could further enhance population-level screening efficiency and enable tiered referral systems for confirmatory diagnostics. Importantly, this approach aligns with resource-sensitive implementation models by prioritizing affordability, accessibility, and adaptability to decentralized healthcare settings. Thus, the integration of immune checkpoint associated biomarker profiling into public health frameworks represents a critical translational axis linking molecular neuroimmunology to actionable early detection strategies in LMIC contexts (Table 2).
Immune Checkpoint Signaling in the Aging Brain: Implications for Early Alzheimer’s Disease Pathogenesis
| Immune Checkpoint | CNS Cell Types Involved | Mechanisms in AD Pathogenesis | Evidence in Early/Asymptomatic AD | Cross-talk with Neuropathology | Therapeutic Implications |
|---|---|---|---|---|---|
| PD-1 (Programmed cell death protein-1) | Microglia, infiltrating CD8+ and CD4+ T cells | PD-1 upregulation promotes T cell exhaustion and dampens neuroprotective inflammation; restricts microglial phagocytosis | Elevated PD-1 expression observed in aged murine models and preclinical AD brains | Impairs clearance of amyloid-β and enhances tau-induced synaptic loss | Anti–PD-1 blockade shows potential in restoring immune surveillance and reducing pathology in murine AD models |
| CTLA-4 (Cytotoxic T lymphocyte-associated protein 4) | Regulatory T cells (Tregs), astrocytes (via indirect signaling) | Suppresses effector T cell activation and alters cytokine milieu in the aging CNS | Early-stage AD shows increased CTLA-4+ Tregs correlating with reduced neuroinflammation | Linked with increased IL-10 and TGF-β secretion, potentially protecting against early neuronal damage | Modulating CTLA-4 may fine-tune Treg function without inducing neurotoxicity |
| LAG-3 (Lymphocyte-activation gene 3) | T cells, microglia, possibly neurons | Inhibits TCR signaling, induces immune senescence, and suppresses antigen-presenting cell (APC) function in microglia | LAG-3 expression upregulated in MCI and early AD patients; correlates with cognitive decline | Interacts with α-synuclein and tau aggregates; impairs synaptic homeostasis | LAG-3 inhibition may restore immune plasticity and support antigen clearance in aging CNS |
| TIM-3 (T cell immunoglobulin and mucin-domain containing-3) | CD4+ T cells, CD8+ T cells, astrocytes | Promotes T cell exhaustion and inhibits IFN-γ production, affecting innate neuroimmune tone | Detected in hippocampal regions of early AD brains; co-expressed with exhausted T cells | May exacerbate neuronal stress responses via glial-immune interactions | Blocking TIM-3 may reinvigorate T cell-mediated neuroprotection in prodromal AD |
| General Dysregulation Trend | Microglia, astrocytes, peripheral lymphocytes | Aging CNS exhibits reduced antigen clearance, increased immune tolerance, and blunted cytokine responsiveness | Immune checkpoint molecules are upregulated before plaque deposition or overt neurodegeneration | Synergistic immune and proteinopathy dysfunction accelerates disease onset | Multi-checkpoint modulation holds promise for early intervention and delaying cognitive decline |
Mechanisms Linking Neuroimmune Dysfunction and Cognitive Decline
The intricate interplay between neuroimmune dysregulation and cognitive decline has emerged as a central axis in the pathogenesis of Alzheimer’s Disease (AD) and age-associated neurodegeneration. While classically considered a neurocentric disorder, compelling evidence now implicates peripheral and central immune pathways including chronic inflammation, immune exhaustion, and checkpoint signaling in accelerating neuronal dysfunction and cognitive impairment. These immunological alterations are neither incidental nor reactive but may constitute upstream pathogenic events shaping the disease trajectory from asymptomatic stages to overt dementia61,62.
To mechanistically contextualize the multi-dimensional interplay between central and peripheral immune systems in driving neurodegeneration, Figure 2 illustrates an integrated framework linking immune checkpoint dysregulation, glial activation, blood–brain barrier disruption, and gut–brain axis perturbations to progressive neuronal dysfunction and cognitive decline in Alzheimer’s Disease.
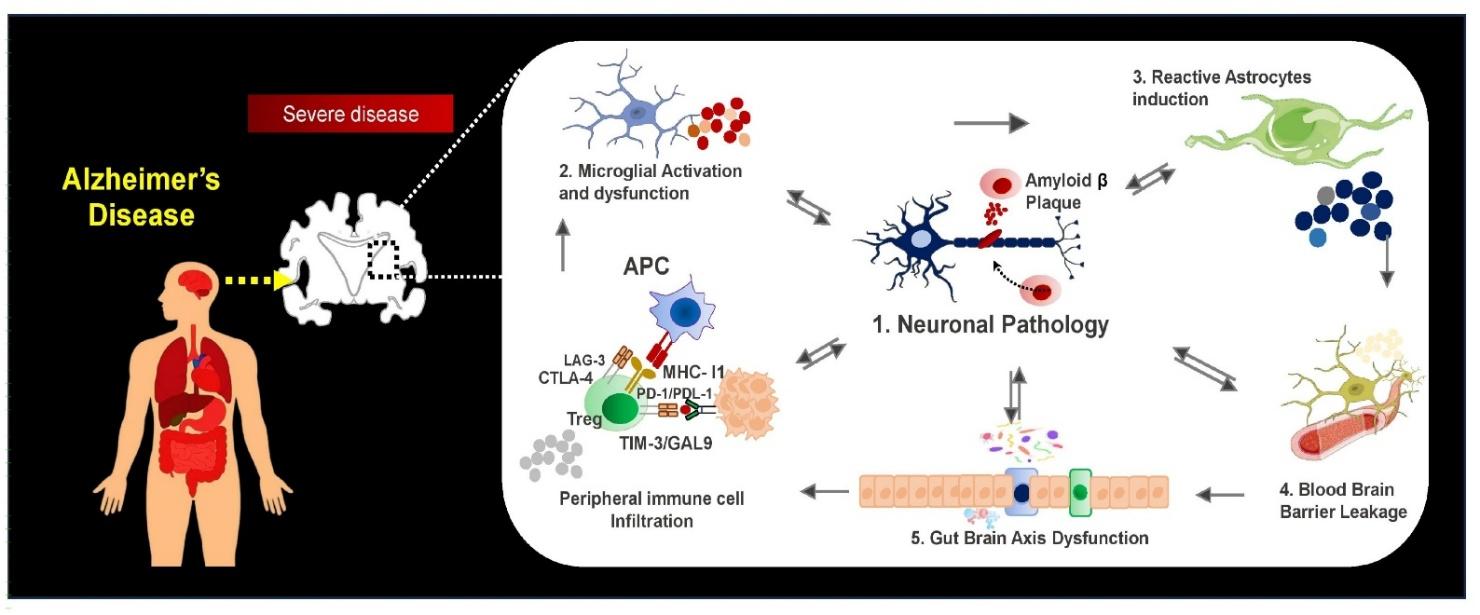
Integrated neuroimmune mechanisms linking systemic immune dysregulation to neuronal pathology in Alzheimer’s Disease. Schematic illustrating the interplay between amyloid-β–driven neuronal pathology, microglial and astrocyte activation, blood–brain barrier disruption, and gut–brain axis dysfunction. Peripheral immune cell infiltration and immune checkpoint signaling (PD-1/PD-L1, CTLA-4, LAG-3, TIM-3) collectively sustain chronic neuroinflammation, driving progressive neuronal injury and cognitive decline. Abbreviations: AD: Alzheimer’s Disease; Aβ: Amyloid-beta; APC: Antigen-presenting cell; PD-1: Programmed cell death protein 1; PD-L1: Programmed death-ligand 1; CTLA-4: Cytotoxic T-lymphocyte-associated protein 4; LAG-3: Lymphocyte activation gene-3; TIM-3: T-cell immunoglobulin and mucin-domain containing-3; CNS: Central nervous system.
Neuroinflammation and Immune Exhaustion in the Aging Brain
Neuroinflammation is a hallmark of the aged central nervous system (CNS) and a well-documented contributor to cognitive decline. With advancing age, glial cells particularly microglia and astrocytes exhibit a shift toward a pro-inflammatory phenotype characterized by increased expression of IL-1β, TNF-α, and IL-6, along with reduced neurotrophic support63. While acute inflammation may serve protective functions, chronic low-level neuroinflammation contributes to synaptic dysfunction, impaired neurogenesis, and ultimately neuronal loss. Concurrently, T cell populations within the CNS and its border tissues undergo phenotypic and functional alterations. Chronic antigenic stimulation leads to T cell exhaustion, marked by upregulation of inhibitory receptors such as PD-1, CTLA-4, LAG-3, and TIM-3. Exhausted T cells fail to clear amyloidogenic substrates or provide adequate cytokine-mediated support, thereby exacerbating glial activation and synaptic damage64. Notably, exhausted PD-1+ CD8+ T cells have been detected in both human AD brains and murine models, spatially correlating with amyloid-β plaques and tau pathology65. The resulting state of immunoparalysis creates a permissive environment for sustained neuropathology, rendering the aging brain less capable of adaptive neuroimmune responses.
Immunosenescence, Inflammaging, and Alzheimer’s Disease Risk
Immunosenescence the age-related decline in immune competence represents a major vulnerability factor in late-life neurodegenerative disease. This process involves thymic involution, reduced naïve T cell output, expansion of oligoclonal memory T cells, and myeloid skewing of hematopoiesis66. These shifts result in impaired pathogen defense and tolerance breakdown, with implications for chronic CNS inflammation and loss of synaptic integrity. In parallel, the phenomenon of "inflammaging" a state of persistent, sterile, low-grade systemic inflammation is increasingly recognized as a driver of AD risk. This process is fueled by cellular senescence, mitochondrial dysfunction, microbial translocation, and dysbiosis. Senescent cells secrete pro-inflammatory factors via the senescence-associated secretory phenotype (SASP), including IL-8, MCP-1, and matrix metalloproteinases, which can cross a compromised blood-brain barrier (BBB) and activate CNS-resident immune cells67. Elevated serum levels of CRP, IL-6, and TNF-α in older adults correlate with accelerated cognitive decline, even in the absence of frank neurodegeneration68. Furthermore, chronic inflammatory states potentiate amyloidogenesis by upregulating β-secretase (BACE1) and impairing amyloid clearance by microglia69. Inflammaging may also promote tau hyperphosphorylation via activation of p38 MAPK and NF-κB pathways70. These molecular cascades reinforce the neurodegenerative loop and underscore the importance of systemic immune health in cognitive aging.
Dysfunctional Phagocytosis, Synaptic Pruning, and Checkpoint-Mediated Immune Suppression
Microglia, the brain’s resident immune cells, play a dual role in synaptic pruning and pathological protein clearance. Under homeostatic conditions, they are crucial for remodeling neural circuits via complement-dependent phagocytosis. However, aging and chronic immune activation lead to a shift toward a dysfunctional, dystrophic microglial phenotype characterized by impaired motility, oxidative stress, and loss of phagocytic competence71. In AD, microglia fail to effectively clear amyloid-β and exhibit reduced expression of key scavenger receptors such as TREM2, CD36, and MERTK72. The accumulation of debris and toxic aggregates drives further microglial activation, cytokine release, and neuronal injury, creating a feed-forward cycle of neurodegeneration. Immune checkpoint signaling compounds this dysfunction. Upregulation of PD-1, LAG-3, and TIM-3 in CNS-infiltrating lymphocytes and resident glia imposes an immunosuppressive tone, inhibiting effective antigen presentation and cytokine crosstalk73. Checkpoint ligation inhibits co-stimulatory signaling, thereby paralyzing both adaptive and innate immune functions in the brain. These pathways, which evolved to maintain immune tolerance and prevent autoimmunity, become maladaptive in the aging CNS, promoting immune evasion by pathogenic protein aggregates and fostering an environment of permissive neurodegeneration.
Environmental Stressors, Infections, and Epigenetic Aging
Environmental and infectious exposures critically modulate neuroimmune tone across the lifespan. Epidemiological studies have linked air pollution (e.g., PM2.5), heavy metals, pesticide exposure, and psychosocial stress with increased risk of cognitive decline and AD. These agents activate innate immune pathways via pattern recognition receptors (e.g., TLR4, NLRP3), triggering inflammasome activation, BBB disruption, and glial priming74,75. Viral infections, particularly herpesviruses (HSV-1, HHV-6), have been proposed as latent accelerants of AD pathology. Reactivation in immunosenescent individuals may stimulate localized inflammation and tau phosphorylation76. Furthermore, COVID-19 has been implicated in long-term cognitive dysfunction, potentially through persistent microglial activation, cerebrovascular damage, and immune dysregulation in genetically vulnerable individuals77. At the molecular level, these stressors may converge on the epigenetic architecture of neuroimmune cells. Age-related changes in DNA methylation, histone modifications, and chromatin remodeling particularly at immune gene loci have been shown to reprogram glial and lymphoid cell identity, biasing toward a pro-inflammatory, dysfunctional state78. For instance, reduced expression of TET2 in microglia has been associated with impaired amyloid clearance, while age-associated methylation drift in T cell regulatory genes potentiates checkpoint overexpression and exhaustion79. Together, these mechanistic insights underscore the multifactorial nature of neuroimmune dysfunction in aging and Alzheimer’s Disease. The convergence of neuroinflammation, immune exhaustion, impaired phagocytic capacity, and environmental insults defines a pathological landscape wherein the immune system not only fails to protect the brain but actively contributes to its decline. Therapeutic strategies aimed at restoring immune surveillance, modulating checkpoint activity, and reversing inflammaging may hold the key to intercepting neurodegeneration at its earliest, most reversible stages80,81.
To delineate the mechanistic underpinnings of neuroimmune alterations across the continuum of preclinical Alzheimer’s Disease, Table 3 provides a qualitative synthesis of key pathways—including neuroinflammation, inflammaging, phagocytic dysfunction, and environmental–epigenetic stressors highlighting their progressive impact on immune homeostasis and cognitive decline.
Mechanistic Pathways Linking Neuroimmune Dysfunction to Cognitive Decline
| Pathway | Key Cellular Components | Mechanistic Features | Stage-wise Progression | Implication in AD Pathogenesis |
|---|---|---|---|---|
| Neuroinflammation | Microglia, astrocytes, peripheral immune cells | Chronic activation of glial cells with increased pro-inflammatory cytokine production | Initiates in aging and amplifies through MCI to AD | Sustained inflammation contributes to synaptic dysfunction and neuronal injury |
| Inflammaging | Senescent immune cells, systemic inflammatory mediators | Low-grade, chronic systemic inflammation driven by aging-associated secretory phenotype (SASP) | Present in healthy aging; progressively exacerbated in MCI and AD | Creates a pro-inflammatory milieu that accelerates neurodegenerative processes |
| Phagocytic Dysfunction | Microglia, astrocytes | Impaired clearance of amyloid-β, cellular debris, and misfolded proteins | Gradual decline in phagocytic efficiency from aging to AD | Promotes accumulation of toxic aggregates and exacerbates neurodegeneration |
| Environmental and Epigenetic Stressors | Immune cells, glial cells | Effects of metabolic stress, pollutants, infections, and epigenetic remodeling of immune-related genes | Cumulative burden increases across lifespan and disease stages | Acts as a trigger and amplifier of neuroimmune dysregulation and vulnerability to AD |
Biomarkers of Neuroimmune Dysregulation in Preclinical Alzheimer’s Disease
The identification of early, reliable biomarkers of neuroimmune dysregulation is critical for intercepting Alzheimer’s Disease (AD) at its preclinical stage prior to irreversible neurodegeneration. Mounting evidence suggests that immunological alterations precede cognitive symptoms by years, if not decades. A growing arsenal of soluble, imaging, and multi-omic biomarkers now enables characterization of neuroimmune states across the disease continuum, with translational potential for both population-level screening and precision-targeted interventions Table 4.
Soluble Immune Checkpoints and Inflammatory Biomarkers
Several soluble immune modulators have emerged as promising indicators of early CNS immune dysfunction. Soluble programmed death ligand-1 (sPD-L1), a circulating form of the immune checkpoint regulator PD-L1, has been detected at elevated levels in individuals with mild cognitive impairment (MCI) and early AD, reflecting systemic immune suppression and T cell exhaustion82. sPD-L1 may also reflect blood-brain barrier (BBB) permeability changes, allowing CNS-derived proteins to enter circulation. Similarly, soluble CD14 (sCD14) a monocyte activation marker has been shown to increase in cognitively normal elderly individuals who later progress to AD83. Elevated plasma levels of sCD14 suggest microglial priming or systemic endotoxin exposure, both of which may trigger innate immune cascades contributing to neuroinflammation. YKL-40 (chitinase-3-like protein 1), predominantly produced by astrocytes and microglia, is elevated in cerebrospinal fluid (CSF) during preclinical and prodromal stages, serving as a robust marker of glial activation84. Among cytokines, interleukin-6 (IL-6) has shown consistent association with longitudinal cognitive decline. IL-6, often upregulated in inflammaging, modulates synaptic plasticity, BBB integrity, and tau phosphorylation85. Notably, elevated IL-6 in CSF and plasma precedes hippocampal atrophy and memory decline in at-risk older adults, indicating its utility as both a mechanistic and prognostic biomarker.
Neuroimaging Correlates of Neuroimmune Activation
Advanced neuroimaging modalities offer spatially resolved, noninvasive insights into neuroinflammation and glial status. Positron emission tomography (PET) ligands targeting the 18-kDa translocation protein (TSPO), a marker of activated microglia, have demonstrated increased binding in the hippocampus, entorhinal cortex, and temporal lobe of individuals with subjective cognitive decline or MCI86. First- and second-generation TSPO ligands, including [11C]PK11195 and [18F]DPA-714, correlate with inflammatory cytokine levels and cognitive impairment, despite the presence of polymorphisms affecting binding affinity. Structural and functional MRI can also infer neuroimmune status through secondary markers. Increased cortical thickness asymmetry, altered functional connectivity in default mode networks, and hyperintensities on T2-weighted imaging have been associated with low-grade inflammation in preclinical AD cohorts87. Moreover, diffusion-weighted MRI has captured microglial activation–associated changes in white matter integrity, supporting the integration of neuroimmune imaging into early AD diagnostics (Table 4).
Peripheral Immune Readouts and Multi-Omic Biomarkers
Peripheral immune signatures especially those derived from blood-based assays are increasingly viable as surrogate markers of CNS immune dysfunction. Flow cytometry analyses have revealed altered frequencies of regulatory T cells (Tregs), exhausted CD8 T cells (PD-1), and pro-inflammatory monocytes in blood samples from individuals in preclinical and prodromal stages of AD88. These shifts mirror CNS pathology and suggest that systemic immune remodeling is tightly linked to early neurodegeneration. Transcriptomic profiling has further identified differentially expressed immune-related genes including those encoding chemokines (e.g., CCL2, CXCL10), interferon response elements, and checkpoint regulators in peripheral blood mononuclear cells (PBMCs) of cognitively normal individuals with amyloid-positive PET scans89. Multi-omic integration of proteomics, epigenomics, and metabolomics has expanded this landscape, revealing distinct immune-metabolic modules associated with silent neurodegenerative processes90. Emerging evidence also supports the use of cell-free nucleic acids (e.g., cfDNA methylation patterns), exosomal miRNAs, and plasma proteome signatures as early indicators of neuroinflammatory dysregulation91. These markers hold promise for minimally invasive screening strategies, particularly in large, aging populations with resource constraints.
Feasibility of Population Screening: Sensitivity, Specificity, and Cost Considerations
To achieve clinical utility, biomarker platforms must balance sensitivity, specificity, scalability, and cost-effectiveness. While cerebrospinal fluid (CSF) biomarkers and TSPO-PET imaging provide high diagnostic specificity for neuroinflammation, their invasive nature and high infrastructural requirements significantly limit their applicability for population-scale screening, particularly in resource-constrained healthcare settings. In contrast, plasma-based biomarkers such as soluble PD-L1 (sPD-L1), interleukin-6 (IL-6), and YKL-40 represent more scalable and cost-efficient alternatives, demonstrating moderate-to-high sensitivity in the range of 70–85% for predicting conversion from cognitively normal states to mild cognitive impairment (MCI) in recent cohort studies92. Recent advances have increasingly focused on composite biomarker strategies that integrate multiple diagnostic dimensions, including plasma protein signatures, genetic risk profiling (e.g., APOE ε4 status), peripheral immune cell phenotyping, and structural neuroimaging metrics. Such multimodal frameworks have been shown to enhance diagnostic accuracy, improve early risk stratification, and enable longitudinal monitoring of at-risk individuals across disease trajectories93. Importantly, these integrated approaches also enhance the robustness of predictive modeling by compensating for the limitations of any single biomarker class.
From a translational and health systems perspective, cost-benefit considerations strongly support the value of early identification strategies targeting immune-activated, amyloid-positive individuals. Although precise economic outcomes vary depending on healthcare infrastructure, diagnostic accessibility, and treatment availability, modeling-based evidence consistently suggests that earlier detection and stratification can reduce long-term healthcare burden by delaying progression to advanced dementia stages, thereby lowering institutional care costs and optimizing intervention timing. These benefits are particularly relevant in systems where late-stage dementia care imposes disproportionate economic strain. Efforts to standardize assay platforms and validate biomarker performance across ethnically and geographically diverse populations remain essential to ensure global applicability, especially given disparities in access to advanced diagnostic technologies and longitudinal cohort data. Nonetheless, the expanding repertoire of immune-linked and plasma-based biomarkers provides a promising and increasingly feasible framework for preclinical Alzheimer’s Disease risk stratification before irreversible neurodegenerative pathology becomes established.
Biomarkers of Neuroimmune Dysregulation in Preclinical Alzheimer’s Disease: A Multimodal Classification with Mechanistic Relevance and Clinical Utility
| Biomarker Category | Representative Markers | Mechanistic Insight | Stage Detected | Sample Type | Sensitivity/Specificity | Limitations | Key References |
|---|---|---|---|---|---|---|---|
| Soluble Immune Checkpoints | sPD-L1, sPD-1, sTIM-3 | Reflects systemic T cell exhaustion, peripheral immune suppression, potential BBB disruption | Preclinical–MCI | Plasma, CSF | Sensitivity ~75%, Specificity ~70% in longitudinal cohorts | Variability due to systemic inflammation, limited standardization | |
| Innate Activation Markers | sCD14, sCD163, YKL-40 | Indicates monocyte/microglial priming, neuroinflammation, and glial activation | Preclinical | Plasma, CSF | Sensitivity 78–85% in predicting progression from CN to MCI | Elevated in other inflammatory states; CSF access invasive | |
| Cytokine and Chemokine Panels | IL-6, TNF-α, IL-8, CCL2, CXCL10 | Signaling network of inflammaging, synaptic remodeling, and leukocyte recruitment | Asymptomatic–MCI | Plasma, serum, CSF | Specificity ~68–80% when combined with APOE status | Highly dynamic; influenced by infections, stress, and age | |
| CSF-Derived Neuroimmune Biomarkers | YKL-40, GFAP, IL-6, IL-10 | Real-time indicators of glial reactivity and CNS inflammation | Preclinical–prodromal | CSF | Specificity >85% when paired with Aβ/tau markers | Invasive collection, regional disparities in access | |
| TSPO PET Imaging | [11C]PK11195, [18F]DPA-714, [11C]PBR28 | Visualizes activated microglia and astrocytes via mitochondrial TSPO binding | Early neuroinflammatory response | Brain (PET scan) | Regional uptake correlates with Braak stage; sensitivity ~80% | Affected by TSPO gene polymorphisms; limited availability | |
| MRI-Based Neuroinflammatory Proxies | T2 FLAIR, DTI, cortical thinning, resting-state connectivity | Structural and functional correlates of glial activation, neurovascular injury | Asymptomatic–early symptomatic | Brain (MRI) | Moderate sensitivity, high accessibility; specific for atrophy | Indirect measure of inflammation; limited molecular specificity | |
| Peripheral Immune Profiling | PD-1+ CD8+ T cells, Tregs, CD14+ monocytes | Reflects systemic immune senescence, checkpoint dysregulation, CNS trafficking potential | Cognitively normal, high-risk individuals | PBMCs (flow cytometry) | Sensitivity ~72%, improves with combinatorial panels | Requires standardization, time-sensitive analysis | |
| Transcriptomic Signatures | Immune gene panels (e.g., IFN-response, MHC-II, cytokine modulators) | Captures immune cell state, exhaustion, and early neuroinflammatory activation | Preclinical–MCI | Whole blood, PBMC RNA | Moderate sensitivity; high for subtype stratification | Requires advanced analysis, influenced by diurnal and lifestyle factors | |
| Multi-omic Composite Panels | Proteomics + Epigenetics + Immune profiling + Imaging | Integrates immune aging, metabolism, and neuroinflammation for robust risk prediction | Asymptomatic–prodromal | Plasma + CSF + Imaging | Sensitivity >90% in recent multimodal ADNI analyses | Complex logistics; requires interdisciplinary infrastructure | |
| Exosomal & Cell-free Biomarkers | Exo-miR-146a, exo-IL-6, cfDNA methylation of immune genes | Reflects CNS-origin immune communication and epigenetic dysregulation | Preclinical | Plasma, serum | High specificity (>85%) in exosomal RNA; cfDNA still emerging | Technical variability; early-phase validation |
Translational Therapeutics and Immunoprevention in Alzheimer’s Disease
As the immunological underpinnings of Alzheimer’s Disease (AD) become increasingly defined, immunotherapeutic and immune-preventive strategies are rapidly emerging as next-generation tools to combat neurodegeneration. While most historical approaches have targeted amyloid-β and tau directly, newer interventions aim to restore immune homeostasis, enhance glial resilience, and reverse maladaptive Immunosenescence. This paradigm shift opens novel translational avenues that bridge oncology, immunology, and neuroscience, offering hope for disease interception during the preclinical or prodromal stages of AD.
Immune-Based Clinical Trials in Alzheimer’s Disease
Checkpoint blockade and innate immune modulation, long pillars of cancer immunotherapy, are now being evaluated for their potential to rejuvenate neuroimmune function in AD. Early-phase trials using immune checkpoint inhibitors such as anti–PD-1 or anti–PD-L1 antibodies have demonstrated the capacity to enhance clearance of amyloid-β plaques and re-activate exhausted T cells in murine models of AD115,115. Systemic PD-1 blockade has been shown to trigger monocyte infiltration and transient immune cell reprogramming within the CNS, resulting in cognitive improvements in transgenic mice. However, translation to human trials remains cautious, given risks of autoimmunity and neuroinflammation. TREM2 (Triggering Receptor Expressed on Myeloid cells 2) has also become a central target. As a microglial activation receptor involved in phagocytosis and inflammatory regulation, TREM2’s modulation holds promise for promoting beneficial microglial responses while limiting chronic inflammation. Agonistic anti-TREM2 antibodies such as AL002 are currently in Phase 2 trials (e.g., INFRONT-AD), aiming to activate microglial-mediated clearance of amyloid and tau aggregates while preserving synaptic function116. Preliminary human data show target engagement and CNS penetration, though clinical efficacy outcomes are pending. Other immunotherapeutic strategies in development include vaccines targeting amyloid, tau, or inflammatory peptides. UB-311, an epitope-specific anti-Aβ vaccine, has shown safety and immunogenicity in Phase 2 trials, with reduced amyloid accumulation and preserved cognition in a subset of patients117. These immunizations are designed to induce a regulatory rather than cytotoxic immune response, circumventing the adverse encephalitic reactions observed in earlier vaccine programs like AN1792. Collectively, these trials represent a maturing pipeline of immune-directed therapies that extend beyond amyloid-centric paradigms.
Preclinical Evidence for Immune Reprogramming
The concept of immune rejuvenation restoring youthful immune function and plasticity is gaining traction as a therapeutic strategy in AD. Several preclinical studies demonstrate that Senolytics agents, which selectively eliminate senescent cells, can ameliorate neuroinflammation and enhance cognitive performance in aged mice. The dasatinib-quercetin combination has been particularly effective in clearing senescent glial cells and reducing SASP cytokine levels within the hippocampus118. Similarly, mTOR (mechanistic target of rapamycin) inhibitors such as rapamycin and everolimus have shown capacity to dampen microglial activation, enhance autophagic clearance of misfolded proteins, and improve synaptic plasticity in AD models119. Chronic mTOR activation is a hallmark of Immunosenescence and contributes to diminished T cell function and glial dysregulation. Pharmacological modulation of mTOR may restore the metabolic flexibility of immune cells, enabling better response to homeostatic and pathogenic signals. HDAC inhibitors, BET bromodomain blockers, and metabolic reprogrammes like NAD+ precursors (e.g., nicotinamide riboside) have also been investigated for their immunomodulatory effects in aging and neurodegeneration. These agents alter epigenetic landscapes and transcriptional profiles of both central and peripheral immune cells, suggesting that re-educating immune cell identity may reverse pro-inflammatory phenotypes and promote tissue regeneration120.
Repurposing Cancer Immunotherapy for Neurodegeneration
There is growing interest in adapting immuno-oncology frameworks for neurodegenerative conditions, particularly given shared features such as immune evasion, cellular exhaustion, and compartmentalized inflammatory signaling. Immune checkpoint modulation, myeloid-targeted strategies (e.g., anti-CSF1R antibodies), and adoptive T cell–based approaches are currently under conceptual and preclinical investigation in the context of neurodegeneration121. In principle, strategies aimed at redirecting cytotoxic lymphocytes or engineered microglia toward pathological substrates mirror approaches used in oncology, although their application in the central nervous system requires substantially greater precision due to the heightened sensitivity of neural tissue and the tightly regulated immune environment of the brain.
Emerging therapeutic concepts also include low-dose or partial immune checkpoint modulation, rather than complete blockade, with the aim of cautiously recalibrating immune tone in aged individuals at risk of Alzheimer’s Disease. Such graded modulation may potentially restore impaired phagocytic activity and T cell surveillance while minimizing the risk of excessive immune activation. However, the therapeutic window for such interventions remains narrow, and their safety profile in neurodegenerative contexts is not yet established. Similarly, cancer-derived neoantigen-targeting paradigms are being conceptually extended to neurodegeneration, where aberrant post-translationally modified proteins may serve as potential immunogenic substrates for selective immune clearance strategies122.
Importantly, despite these promising translational parallels, significant biological and safety constraints must be acknowledged. The central nervous system presents a uniquely sensitive immunological environment, and inappropriate immune activation may lead to neurotoxicity, disruption of neuronal networks, and exacerbation of chronic neuroinflammation. Therefore, while immuno-oncology–inspired approaches provide a valuable conceptual framework, their translation to neurodegenerative diseases will require rigorous validation, careful dose optimization, and strategies ensuring spatial and temporal control of immune activation.
Integrative Approaches: Nutrition, Microbiota, and Lifestyle-Linked Immunomodulation
Beyond pharmacological strategies, integrative interventions that modulate systemic immunity and gut-brain axis dynamics hold substantial promise in immunoprevention. Dietary components such as polyphenols, omega-3 fatty acids, and flavonoids have been shown to downregulate NF-κB and NLRP3 pathways while enhancing regulatory T cell function123. Long-term Mediterranean-style diets are associated with reduced AD risk, partly through attenuation of systemic inflammation and improved gut microbiota diversity. The gut microbiota plays a pivotal role in shaping CNS immunity through microbial metabolites such as short-chain fatty acids (SCFAs), tryptophan catabolites, and endotoxins. Dysbiosis in aging has been linked to microglial priming, increased permeability of the gut-brain barrier, and upregulation of peripheral pro-inflammatory mediators124. Probiotic and prebiotic interventions, as well as fecal microbiota transplantation (FMT), are under active investigation as strategies to rebalance gut-immune interactions and delay cognitive decline. Finally, structured exercise, caloric modulation, and sleep hygiene exert potent immunomodulatory effects. Regular aerobic activity improves monocyte/macrophage polarization, lowers systemic IL-6, and enhances glymphatic clearance of neurotoxins125. Sleep deprivation, conversely, amplifies microglial activation and primes the CNS for inflammatory insults. Integrating lifestyle-based interventions into early-stage prevention programs may provide a cost-effective and scalable complement to pharmacological immunotherapy.
Public Health and Implementation Pathways for Neuroimmune Biomarkers in Alzheimer’s Disease
The recognition of immune dysregulation as an upstream driver of neurodegeneration has catalyzed a paradigm shift in Alzheimer’s Disease (AD) management from late-stage therapeutic intervention to early, preventive action. Integrating neuroimmune biomarkers into public health frameworks, particularly within aging populations, presents both unprecedented opportunity and complex implementation challenges. Realizing the translational potential of these tools demands a coordinated strategy across primary care systems, risk stratification infrastructure, global dementia policy, and equitable access in low- and middle-income countries (LMICs).
Neuroimmune Biomarkers in Primary Care and Aging Programs
Primary care represents the frontline of dementia prevention and is uniquely positioned to incorporate immune-based early detection tools. Unlike neuroimaging or cerebrospinal fluid (CSF) diagnostics, many neuroimmune biomarkers such as plasma sPD-L1, YKL-40, and IL-6 are amenable to blood-based assays that can be integrated into routine health check-ups for older adults126. As aging programs increasingly adopt precision health paradigms, these biomarkers can serve as scalable tools for identifying individuals with silent neuroinflammatory activity who are at elevated risk of cognitive decline. Several aging-focused initiatives, such as the WHO’s Integrated Care for Older People (ICOPE) framework, emphasize functional ability and intrinsic capacity assessments at the community level127. Embedding neuroimmune markers into these protocols may enhance early-stage detection and allow for stratified follow-up or referral to memory clinics. Pilot programs in high-income countries (e.g., NHS Health Checks in the UK) could serve as test beds for implementation, followed by adaptation in LMICs. However, standardization of biomarker thresholds, accounting for comorbidities and population-specific immune baselines, remains essential to avoid false positives and overburdening of specialist services. Primary care providers will also require updated training on interpreting immune biomarker profiles within the broader context of brain health and cognitive aging.
Risk Stratification Models and Screening Infrastructure
Effective risk stratification models incorporating neuroimmune data could transform cognitive decline from a reactive to a preventive care challenge. Machine learning–based predictive models that integrate immune biomarkers, APOE ε4 status, polygenic risk scores, and socio-behavioral metrics are already under development128. These tools can help identify individuals in the preclinical or prodromal stages of AD for enrollment into targeted lifestyle, dietary, or immunomodulatory interventions. Large-scale longitudinal studies, such as the UK Biobank, ADNI, and Indian Longitudinal Aging Study (LASI), are crucial for validating such models across diverse populations129. Public health agencies should prioritize cross-cohort biomarker harmonization and real-world effectiveness assessments, ensuring that stratification tools remain generalizable beyond academic settings. National dementia strategies must also ensure that screening programs do not exacerbate existing healthcare disparities. Tiered screening models where low-cost biomarkers (e.g., IL-6, CRP) serve as first-line tests, followed by confirmatory neuroimaging or CSF biomarkers may offer both cost-efficiency and high sensitivity in resource-limited settings130.
Building Brain Health Equity in LMICs and Underserved Populations
AD is poised to disproportionately affect LMICs and socioeconomically marginalized populations, where aging is rapid, diagnostic capacity is limited, and public health infrastructure remains fragmented131. Despite this, global dementia biomarker research remains dominated by cohorts from North America and Western Europe, risking translational misalignment with global need. To address these gaps, equity-cantered implementation pathways must include locally validated reference ranges for neuroimmune markers, capacity-building for community health workers, and low-barrier access to diagnostics. Mobile health (mHealth) platforms, combined with dried blood spot (DBS) biomarker testing, could be leveraged to bring screening directly to underserved elderly individuals in rural and peri-urban regions132. Community-based participatory research (CBPR) models where local populations are engaged as co-designers of implementation strategies are crucial for building trust and uptake. Culturally sensitive approaches, particularly among populations with low dementia literacy or strong stigma, are essential to prevent underdiagnosis and therapeutic nihilism133.
Policy, Ethics, and Global Governance
Incorporating neuroimmune biomarkers into early AD detection programs raises important ethical and governance considerations. The predictive power of these biomarkers, in the absence of widely available curative treatments, challenges traditional risk disclosure protocols. There is a pressing need for public health ethics frameworks that address autonomy, consent, psychological impact, and potential discrimination in insurance or employment134. Global organizations such as the World Health Organization (WHO) and OECD have begun to outline guiding principles. The WHO Global Action Plan on the Public Health Response to Dementia 2017–2025 advocates for the integration of novel diagnostics into primary care pathways while ensuring equitable access and social protection135. Ethical implementation must also include transparency in data sharing, cross-border biomarker regulation, and citizen science engagement. At a national level, health ministries should incorporate neuroimmune biomarkers into dementia registries, electronic health records (EHRs), and policy roadmaps aligned with universal health coverage goals. Intersectoral collaboration with aging, digital health, immunology, and policy domains will be key to building sustainable, actionable brain health frameworks. Neuroimmune biomarkers have the potential to revolutionize public health approaches to Alzheimer’s Disease by enabling early, precision-guided prevention. However, their implementation requires more than scientific validation it demands inclusive policies, infrastructure investments, ethical oversight, and cultural adaptation. The future of dementia care must transition from reactive crisis management to proactive immune-informed prevention, rooted in health equity and global solidarity.
Worldwide Trends in Alzheimer’s Disease: Public Health Implications for Early Diagnosis and Healthy Aging
Alzheimer’s Disease (AD) represents a growing public health crisis, with over 55 million affected individuals globally and projections surpassing 139 million by 2050136. Nearly 70% of cases remain undiagnosed, often progressing silently through preclinical stages, particularly in aging and immunosenescent populations137(Table 5). The disproportionate burden in low- and middle-income countries (LMICs), where over 65% of cases reside, highlights urgent disparities in biomarker access and early detection infrastructure138. Integrating neuroimmune biomarkers and immune checkpoint profiling into routine public health screening could enable timely interventions and reduce the escalating global cost of dementia care, now exceeding $1.4 trillion annually139.
Global Alzheimer’s Disease Statistics Relevant to Public Health, Aging, and Preclinical Detection (2024–2025 Estimates)
| Parameter | Global Estimate (2024–2025) | Public Health Implication | Relevance to Neuroimmune & Preclinical AD Axis |
|---|---|---|---|
| Global AD Cases (all stages) | ~55 million people (WHO, 2024) | Major neurodegenerative epidemic | Highlights need for early neuroimmune diagnostics |
| Projected AD cases by 2050 | >139 million | Rapidly aging global population | Accelerated burden in immune-aging societies |
| Proportion of undiagnosed/preclinical cases | 60–70% remain undiagnosed | Silent progression overwhelms systems | Biomarker-guided neuroimmune detection essential |
| Annual new cases | ~10 million/year | Persistent increase in incidence | Early intervention requires immune-checkpoint-based screening |
| AD-related deaths/year | >2 million globally | Major cause of mortality in elderly | Reflects late diagnosis; preventive biomarkers crucial |
| Average delay from symptom onset to diagnosis | ~2.8 years | Diagnostic delay worsens outcomes | Emphasizes value of immune biomarker panels |
| Global cost of dementia care | ~$1.4 trillion USD (2024) | High socio-economic toll | Public health systems under strain, especially LMICs |
| Cases in Low- and Middle-Income Countries (LMICs) | >65% of global burden | Disproportionate impact | Gaps in neuroimmune infrastructure and access to diagnostics |
| Preclinical AD in asymptomatic older adults (aged >60) | ~30–35% globally (PET/CSF-based) | Silent burden rising | Validates neuroimmune biomarkers in aging surveillance |
| Screening programs with validated immune biomarkers | <5% countries have formal programs | Vast underutilization | Policy gap in integrating neuroimmune diagnostics |
DISCUSSION
The expanding intersection between neuroimmunology and Alzheimer’s Disease (AD) pathogenesis has substantially reshaped prevailing models of neurodegeneration, positioning immune dysregulation as a contributory factor in early disease evolution rather than a purely downstream consequence of protein aggregation150,151. This evolving framework highlights the neuroimmune axis as a critical interface linking aging biology, central nervous system (CNS) homeostasis, and systemic inflammatory processes. However, the extent to which immune alterations act as primary drivers versus secondary amplifiers of pathology remains incompletely resolved, necessitating cautious interpretation of emerging findings151.
A central feature of this paradigm is the progressive emergence of age-associated immune remodeling, often described as “inflammaging,” characterized by chronic low-grade inflammation, immunosenescence, and functional exhaustion of adaptive immune compartments152. Within the CNS, alterations in microglial activation states, impaired phagocytic capacity, and sustained pro-inflammatory signaling contribute to a permissive environment for neurodegenerative processes. Concurrently, dysregulated peripheral immune responses including altered trafficking of T lymphocytes and monocyte-derived cells reinforce bidirectional neuroimmune interactions152,153. Increased expression of inhibitory immune checkpoint molecules such as programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) may further attenuate immune surveillance and limit effective clearance of pathogenic aggregates153. Notably, emerging mechanistic evidence suggests that age-related epigenetic remodeling including DNA methylation changes at immune regulatory loci such as PDCD1 and histone modifications influencing microglial genes like TREM2 may contribute to sustained immune dysfunction and chronic neuroinflammatory signaling, thereby linking Immunosenescence with persistent neurodegenerative vulnerability154.
The increasing focus on neuroimmune biomarkers reflects their potential to capture early pathophysiological alterations preceding clinical symptom onset. Peripheral and central markers including soluble checkpoint molecules, monocyte activation markers, and glial-derived proteins have shown associations with disease progression in observational and translational studies155. However, their clinical applicability remains constrained by several critical limitations. Many immune biomarkers lack disease specificity and may be influenced by comorbid inflammatory conditions, infections, or metabolic disorders. In addition, variability in assay platforms, lack of standardized protocols, and limited reproducibility across cohorts pose significant barriers to clinical translation155,156. These challenges are particularly pronounced in low- and middle-income countries, where access to advanced diagnostics such as cerebrospinal fluid analysis and molecular neuroimaging is limited. Furthermore, high background levels of systemic inflammation in such settings may increase the risk of false-positive interpretations, thereby complicating population-level screening strategies156. Consequently, while neuroimmune biomarkers hold promise for early risk stratification, their integration into routine clinical practice will require rigorous validation, harmonization of methodologies, and cost-effective implementation frameworks.
From a therapeutic perspective, the shift toward immune-directed strategies represents a conceptual departure from traditional amyloid-centric approaches. Monoclonal antibodies targeting amyloid-β, including Aducanumab and Lecanemab, have demonstrated reductions in amyloid burden but have yielded modest and sometimes inconsistent clinical benefits, and are associated with adverse effects such as amyloid-related imaging abnormalities (ARIA)157. These limitations underscore the need for alternative or complementary strategies. In contrast, immune-modulatory approaches including checkpoint pathway modulation, microglial reprogramming via TREM2 signaling, and targeting cellular senescence aim to restore immune homeostasis rather than directly eliminate pathological aggregates. While preclinical findings are promising, these approaches remain in early translational stages, and their safety profiles require careful evaluation given the potential risks of excessive immune activation and the unique immunological constraints of the CNS157.
Beyond pharmacological interventions, broader determinants of immune function including metabolic health, environmental exposures, and microbiome composition are increasingly recognized as modulators of neurodegenerative risk. Interactions between systemic inflammation and epigenetic regulation may influence disease susceptibility across the lifespan, further emphasizing the complexity of neuroimmune dynamics154,156. Importantly, much of the current evidence base is derived from high-income populations, raising concerns regarding generalizability. Variations in environmental exposures, infectious disease burden, and healthcare infrastructure across global settings may shape neuroimmune trajectories differently, underscoring the need for geographically diverse and population-specific research156.
From a public health and translational perspective, the integration of neuroimmune insights into early detection and prevention strategies presents both opportunities and challenges. While multi-omic and immune-based profiling approaches may enhance risk stratification, their implementation raises critical considerations related to cost, accessibility, assay standardization, and ethical implications particularly in the absence of definitive disease-modifying therapies155,156,157. These constraints highlight the importance of aligning scientific advances with health system capacity and policy frameworks to ensure equitable and responsible translation.
In summary, the conceptualization of AD as a disorder involving neuroimmune dysregulation provides a valuable framework for understanding early disease mechanisms and identifying potential intervention points. Nevertheless, substantial gaps remain in defining causal relationships, validating biomarkers across diverse populations, and establishing the long-term safety and efficacy of immune-targeted therapies. Future progress will depend on integrating mechanistic insights with longitudinal human studies and implementation science approaches to determine whether modulation of the neuroimmune axis can meaningfully alter disease trajectories150,151,152,153,154,155,156,157.
FUTURE DIRECTIONS
The reconceptualization of Alzheimer’s Disease (AD) as an immune-mediated disorder offers a powerful foundation for advancing both diagnostic and therapeutic paradigms. Immune checkpoint molecules such as PD-1, PD-L1, CTLA-4, and LAG-3 are emerging as dual-purpose tools: not only as therapeutic targets but also as dynamic biomarkers for tracking disease onset and progression. Their expression profiles in both central and peripheral compartments may reflect evolving immune exhaustion, synaptic dysfunction, and early-stage neuroinflammation enabling multi-dimensional stratification of at-risk individuals158,159 (Figure 3).
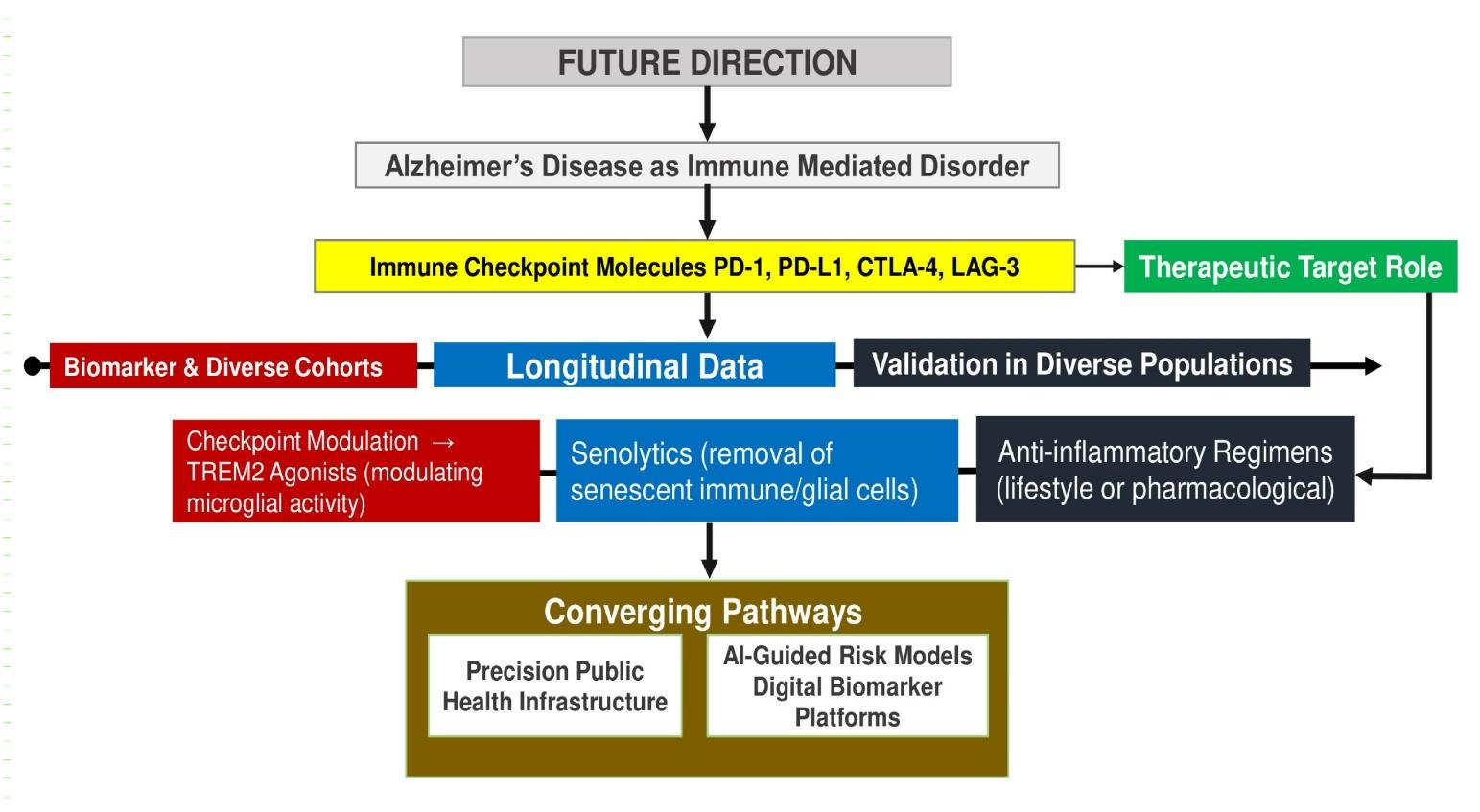
Flowchart of Future Prospects and Emerging Strategies in Immune-Based Alzheimer’s Research. Flowchart highlighting immune checkpoint molecules as dual biomarkers and therapeutic targets, integrating longitudinal validation and emerging strategies (checkpoint modulation, TREM2 targeting, Senolytics, anti-inflammatory approaches) within precision public health and artificial intelligence driven frameworks for early disease interception. Abbreviations: AD: Alzheimer’s Disease; PD-1: Programmed cell death protein 1; PD-L1: Programmed death-ligand 1; CTLA-4: Cytotoxic T-lymphocyte-associated protein 4; LAG-3: Lymphocyte activation gene-3; TREM2: Triggering receptor expressed on myeloid cells 2; AI: Artificial intelligence; LMICs: Low- and middle-income countries.
To fully realize this potential, investment in longitudinal, ethnically diverse cohorts with serial immune, imaging, and cognitive data is critical. Most current biomarker frameworks are derived from predominantly white, high-income populations, limiting translational equity. Building population-specific reference ranges, especially in low- and middle-income countries (LMICs), must become a priority within international dementia research consortia160. The integration of immune biomarkers into digital health platforms and AI-guided risk prediction tools also demands scalable public health infrastructure that bridges geriatric medicine, immunology, and informatics161 (Figure 3). Positioning neuroimmune dysregulation as a modifiable axis reframes cognitive decline not as an inevitable consequence of aging but as an intervenable trajectory. Therapeutic strategies including checkpoint recalibration, TREM2 activation, senolytics clearance, and anti-inflammatory diets have the potential to reprogram immune tone, delay pathology, and extend cognitive health span162. A global call to action must now converge immunology, geriatrics, and public health into a unified front against dementia. This requires cross-sector coordination, ethical frameworks for preclinical detection, and the political will to fund preventive neurology at scale. As immune signatures become the harbingers of neurodegeneration, the future of AD prevention lies in decoding and correcting the language of immune aging166.
CONCLUSION
The evolving understanding of Alzheimer’s Disease (AD) as a disorder influenced by neuroimmune interactions provides an expanded framework for interpreting early disease mechanisms and identifying potential points of intervention. Accumulating evidence suggests that neuroimmune dysregulation including microglial priming, immune checkpoint signaling, and systemic inflammaging may contribute to both the initiation and progression of neurodegenerative processes, although the precise causal relationships remain to be fully established. Advances in biomarker research, including soluble immune mediators, glial activation markers, and neuroinflammation-targeted imaging approaches, offer promising avenues for detecting early pathophysiological changes associated with preclinical AD. However, the clinical applicability of these biomarkers is currently limited by issues related to specificity, reproducibility, and standardization across populations and platforms. Similarly, emerging therapeutic strategies targeting immune pathways such as checkpoint modulation, microglial signaling pathways, and cellular senescence have demonstrated encouraging results in preclinical and early translational studies, but require rigorous clinical validation to establish safety, efficacy, and long-term outcomes. Beyond targeted therapies, broader determinants of immune function, including metabolic health, environmental exposures, and lifestyle factors, may influence neurodegenerative risk and represent additional areas for preventive investigation. Nevertheless, translating these insights into clinical and public health practice remains challenging. Barriers related to cost, infrastructure, and population heterogeneity are particularly relevant in low- and middle-income countries, where the burden of dementia is rapidly increasing and access to advanced diagnostic technologies is limited. In this context, future progress will depend on integrating mechanistic insights from neuroimmunology with longitudinal clinical studies, biomarker validation efforts, and implementation science approaches. A more comprehensive understanding of how immune dysregulation interacts with neurodegenerative pathways may ultimately inform more precise and equitable strategies for early detection and intervention. While the neuroimmune paradigm represents a promising direction, its successful translation into clinical practice will require careful evaluation, interdisciplinary collaboration, and alignment with global health priorities.
ABBREVIATIONS
AA: Accelerated Aging; AD: Alzheimer’s Disease; AUC: Area Under the Curve; CD14: Cluster of Differentiation 14; CNS: Central Nervous System; CRP: C-Reactive Protein; CSF: Cerebrospinal Fluid; CTLA-4: Cytotoxic T-Lymphocyte Antigen 4; DAMPs: Damage-Associated Molecular Patterns; EHR: Electronic Health Record; IFN-γ: Interferon-gamma; IL-6: Interleukin-6; ICPs: Immune Checkpoint Pathways; IS: Immunosenescence; LAG-3: Lymphocyte Activation Gene-3; LMICs: Low- and Middle-Income Countries; mTOR: Mechanistic Target of Rapamycin; MRI: Magnetic Resonance Imaging; NDDs: Neurodegenerative Diseases; NID: Neuroimmune Dysregulation; PD-1: Programmed Cell Death Protein 1; PD-L1: Programmed Death-Ligand 1; PAMPs: Pathogen-Associated Molecular Patterns; PET: Positron Emission Tomography; PHI: Public Health Intervention; PMI: Precision Medicine Initiative; PPH: Precision Public Health; SASP: Senescence-Associated Secretory Phenotype; sCD14: Soluble CD14; sPD-L1: Soluble Programmed Death-Ligand 1; TIM-3: T-cell Immunoglobulin and Mucin-Domain Containing-3; TLRs: Toll-Like Receptors; TME: Tissue Microenvironment; TNF-α: Tumor Necrosis Factor-alpha; TSPO: Translocator Protein (18kDa); WHO: World Health Organization; YKL-40: Chitinase-3-Like Protein 1.
Acknowledgments
None.
Author’s contributions
Dr. Arpita Mukherjee conceived the study, designed the methodology, conducted the research, created all figures and illustrations, analyzed the data, and wrote the manuscript. Dr. Arpita Mukherjee takes full responsibility for the integrity, accuracy, and completeness of the work.
Funding
None.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have not used generative AI (a type of artificial intelligence technology that can produce various types of content including text, imagery, audio and synthetic data).
Competing interests
The authors declare that they have no competing interests.