

The Role of α-2A-Adrenergic Receptors in Modulating Epileptiform Activity and the Therapeutic Potential of Brimonidine (UK14,304)
- Department of Human Anatomy, Federal University of Lafia, Nassarawa, Nigeria
- Faculty of Veterinary Medicine, Usmanu Danfodiyo University, P.M.B 2346, Sokoto, Nigeria
- Department of Human Physiology, Federal University of Lafia, Nigeria
- Department of Imaging, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia
Abstract
Epilepsy remains a profound clinical challenge, with approximately one-third of patients exhibiting resistance to current antiseizure medications (ASMs). This persistent drug resistance highlights the limitations of traditional ion-channel targets and underscores an urgent need for alternative, mechanism-based neuromodulatory strategies. This review synthesizes current findings on the therapeutic potential of the noradrenergic system, specifically the -adrenergic receptor ( -AR), as a modulator of network excitability. We examine the biology of the -AR, which acts as a presynaptic "brake" on glutamatergic transmission, and review preclinical evidence evaluating brimonidine (UK14,304) as a prototype agonist. Emerging data suggest that selective -activation offers a state-dependent antiseizure mechanism, thereby suppressing pathological hypersynchrony while sparing physiological transmission. Finally, we propose that future therapeutic success depends upon the development of biased ligands and focal delivery systems, which may effectively harness -AR signaling to suppress focal seizures and mitigate the risk of Sudden Unexpected Death in Epilepsy (SUDEP).
Introduction
Epilepsy is a chronic neurological disorder characterized by spontaneous, recurrent seizures resulting from abnormal, hypersynchronous neuronal activity 1. Globally, the burden of this condition is profound: approximately 50 million people live with epilepsy, with over 5 million new cases diagnosed annually, predominantly in low- and middle-income countries 2.
To manage this condition, current antiseizure medications (ASMs) primarily target voltage-gated ion channels or modulate GABAergic and glutamatergic transmission to restore the critical balance between excitation and inhibition 3. However, despite the introduction of newer agents, approximately one-third of patients remain drug-resistant, and many others experience adverse effects that limit long-term utility 4,5. These persistent clinical limitations underscore an urgent need to explore alternative molecular systems that modulate epileptogenic circuits.
One promising avenue involves neuromodulatory systems that tune network excitability, such as the noradrenergic system 6. Norepinephrine (NE) exerts powerful, state-dependent effects on cortical and hippocampal synchrony, influencing both seizure threshold and termination 7. Among the receptors mediating these effects, the αA-adrenergic receptor (αA-AR) is particularly relevant 8. Owing to its high expression in limbic and cortical regions, αA-ARs are strategically positioned to regulate glutamatergic output and network synchronization.
Consequently, pharmacological targeting of this receptor represents a promising, though complex, strategy 8. Although activating αA-AR effectively reduces excitability 9, systemic agonists such as clonidine and dexmedetomidine have traditionally caused notable sedation and hypotension 10,11. These side effects have restricted their use in long-term epilepsy treatment. Nonetheless, the therapeutic viability of this target is now being revisited through the lens of modern pharmacology. By using biased ligands that selectively activate anticonvulsant pathways without engaging sedative ones 12, or by applying focal delivery methods to target only the epileptogenic zone 13, it may be possible to capitalize on the strong antiseizure effects of the αA receptor while avoiding systemic side effects 14.
To understand the specific physiological functions and therapeutic possibilities of the αA-AR subtype within the intricate adrenergic signaling landscape, highly selective pharmacological probes are crucial. In this context, brimonidine (UK14,304) is essential to this review. Initially identified as a potent and selective αA-adrenoceptor agonist, it serves as a primary reference compound for studying the pharmacology of αA-ARs 15. It mimics the actions of endogenous norepinephrine at these receptors. As a result, it activates G/o-coupled signaling pathways, which are critical for downstream effects. Activation of these pathways suppresses adenylyl cyclase, lowers cAMP signaling, and modulates ion conductance. Together, these changes decrease neuronal excitability and reduce neurotransmitter release 15. Brimonidine exhibits neuroprotective effects in preclinical models of retinal injury and optic neuropathy, and it has gained clinical approval for ocular indications 16,17. Because of these properties, analyzing its pharmacological profile provides vital insights into the mechanisms of targeted αA-adrenoceptor modulation. Such analysis also informs the translational value of αA-adrenoceptor-focused therapies. In experimental systems, brimonidine's neuroprotective actions include enhanced survival of retinal ganglion cells and reduced excitotoxic damage. However, definitive clinical evidence for neuroprotection in human central nervous system disorders remains limited and inconclusive 18,19. This review aims to comprehensively evaluate current evidence on the pharmacology of αA-adrenoceptors, focusing on the selective agonist brimonidine as a key reference compound. The review also seeks to clarify how αA-adrenoceptor activation affects neuronal excitability and to assess the potential of αA-adrenoceptor modulation as a therapeutic strategy for future drug development.
Methods
Literature Search Strategy and Selection Criteria
This review was conducted as a scoping review utilizing a systematic literature search. This approach was chosen to map the extent, nature, and mechanistic direction of the available evidence regarding αA-adrenergic receptor (αA-AR) signaling and the selective agonist brimonidine (UK14,304) in epilepsy, thereby identifying critical knowledge gaps for future translational work 20,21.
The review question was structured using the Population–Concept–Context (PCC) framework 22:
-
Population: Preclinical central nervous system (CNS) models relevant to epilepsy, including
ex vivo brain-slice preparations,in vivo animal models, and mechanistic receptor-pharmacology studies. -
Concept: Modulation of α2A-AR signaling, with a specific focus on the pharmacological profile of brimonidine (UK14,304).
-
Context: Seizure-relevant biology, encompassing network excitability, glutamatergic/GABAergic transmission, epileptiform activity, seizure-linked neuroprotection, and autonomic mechanisms relevant to Sudden Unexpected Death in Epilepsy (SUDEP).
Search Strategy
Electronic literature searches were conducted in PubMed/MEDLINE, Scopus, and Web of Science from database inception through January 2026. To maximize search sensitivity, the strategy combined controlled vocabulary with free-text keywords and Boolean operators across three primary domains:
-
Receptor Target: “alpha-2A adrenergic receptor,” “α2A-AR,” “ADRA2A,” and related α2A-adrenoceptor terminology.
-
Pharmacological Probe: "Brimonidine," "UK14,304," and related α2A-agonist terms.
-
Mechanistic Context: "epilepsy," "seizure," "epileptiform activity," "network excitability," "SUDEP," "neuromodulation," and "neuroprotection."
Additionally, the reference lists of all eligible articles and relevant seminal reviews were manually screened to capture supplemental studies not identified in the primary database query.
Eligibility Criteria
Eligibility criteria were defined a priori. To be included in the primary synthesis, studies were required to be peer-reviewed, English-language original research articles that met at least one of the following scientific criteria:
-
Ex vivo ,in vivo , or mechanistic preclinical studies evaluating the role of central α2A-AR signaling in network excitability, synaptic transmission, epileptiform bursting, or seizure-linked autonomic dysfunction. -
Studies specifically investigating brimonidine (UK14,304) or closely related α2A-agonists in the context of central neuroprotection, antiseizure efficacy, or mechanisms relevant to epilepsy translation. (Note: Seminal review articles were included solely to provide conceptual background and support reference tracking, rather than serving as primary evidence).
Studies were excluded if they were conference abstracts lacking full text, editorials, commentaries, or non-English publications. To ensure the review captured the most current and highly relevant mechanistic data, preprints were universally eligible for inclusion provided they strictly met the predefined PCC criteria. However, because preprints have not yet undergone formal peer review, their findings were interpreted with appropriate methodological caution. Finally, studies were excluded if they lacked interpretable experimental controls, utilized poorly validated models preventing meaningful mechanistic interpretation, or did not clearly relate to CNS αA-AR biology.
Data Extraction and Synthesis
Following the removal of duplicate records, citations were screened by title and abstract, followed by a full-text review against the eligibility criteria. For each included source, data were charted regarding the study type, experimental model (species/tissue preparation), epileptogenic trigger, pharmacological intervention, comparator condition, and principal mechanistic findings. Particular analytical focus was placed on variables likely to influence pharmacological interpretation, such as the receptor subtype interrogated and whether the observed drug effects reflected global network suppression versus state-dependent modulation of pathological activity.
Due to the substantial heterogeneity in model systems, experimental designs, and endpoints across the selected literature, the evidence was synthesized narratively rather than via meta-analysis. In alignment with standard scoping review methodology, a formal quantitative risk-of-bias tool was not applied 21,22. However, the methodological rigor of the included studies—specifically model validity, pharmacological specificity, and the use of appropriate controls—was assessed qualitatively throughout the synthesis to ensure robust evidence mapping (Figure 1).
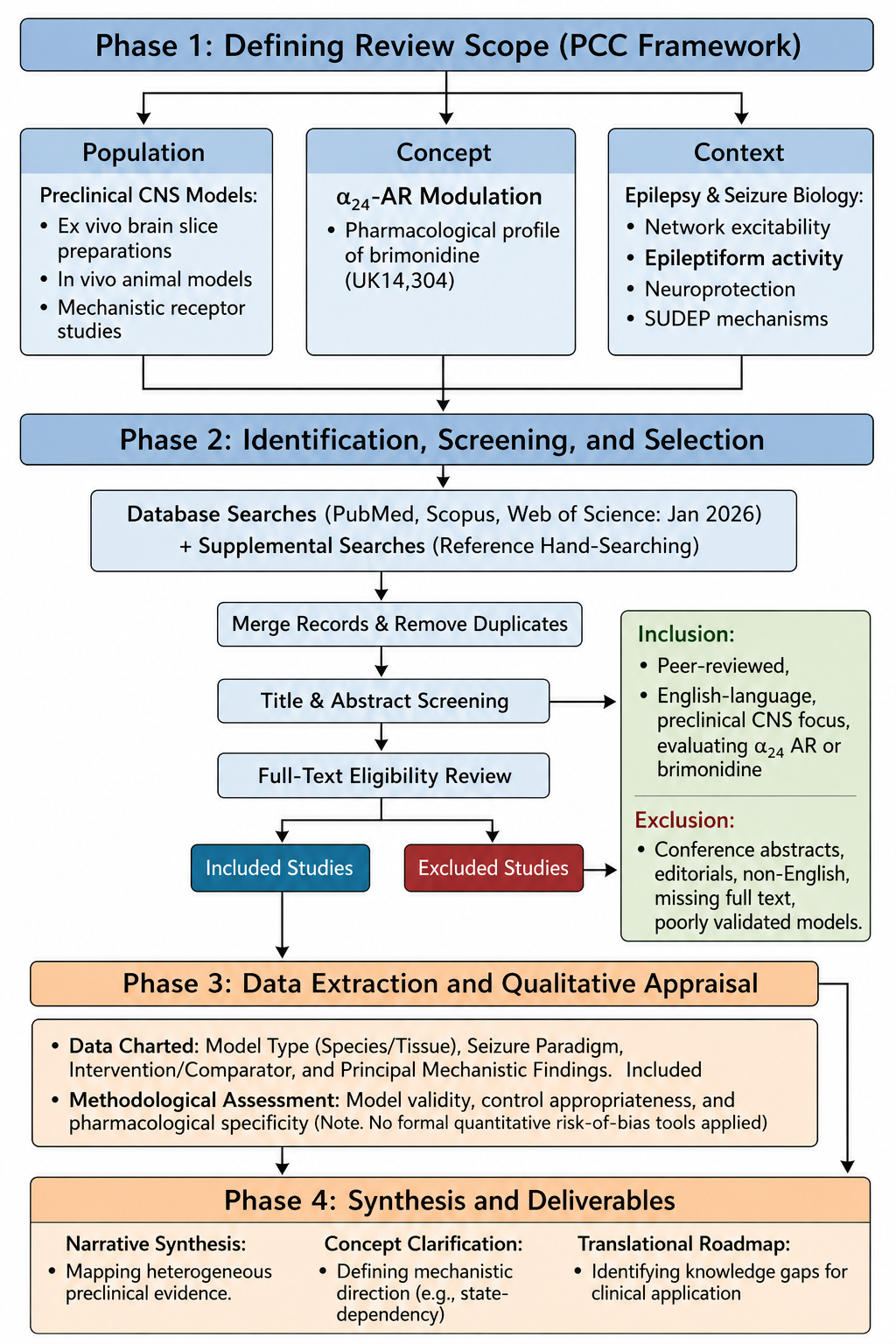
Conceptual framework and methodological workflow of the scoping review. The review process was structured across four distinct phases to map the preclinical literature regarding α2A-adrenergic receptor (α2A-AR) signaling in epilepsy. Phase 1 defines the study parameters using the Population, Concept, Context (PCC) framework. Phase 2 illustrates the literature identification, screening, and selection pipeline, including database queries, deduplication, and the application of predefined eligibility criteria (with an explicit, predefined inclusion for a highly relevant mechanistic preprint). Phase 3 outlines the data extraction parameters and qualitative methodological appraisal, conducted without formal quantitative risk-of-bias tools. Phase 4 summarizes the final deliverables, culminating in a narrative synthesis, concept clarification, and a translational roadmap for future clinical applications.
The selection process and the number of records identified, screened, and included are detailed in the PRISMA-ScR flow diagram (Figure 2). Furthermore, the characteristics, models, and principal findings of the included preclinical studies are charted in Table 1 to support the data extraction process.
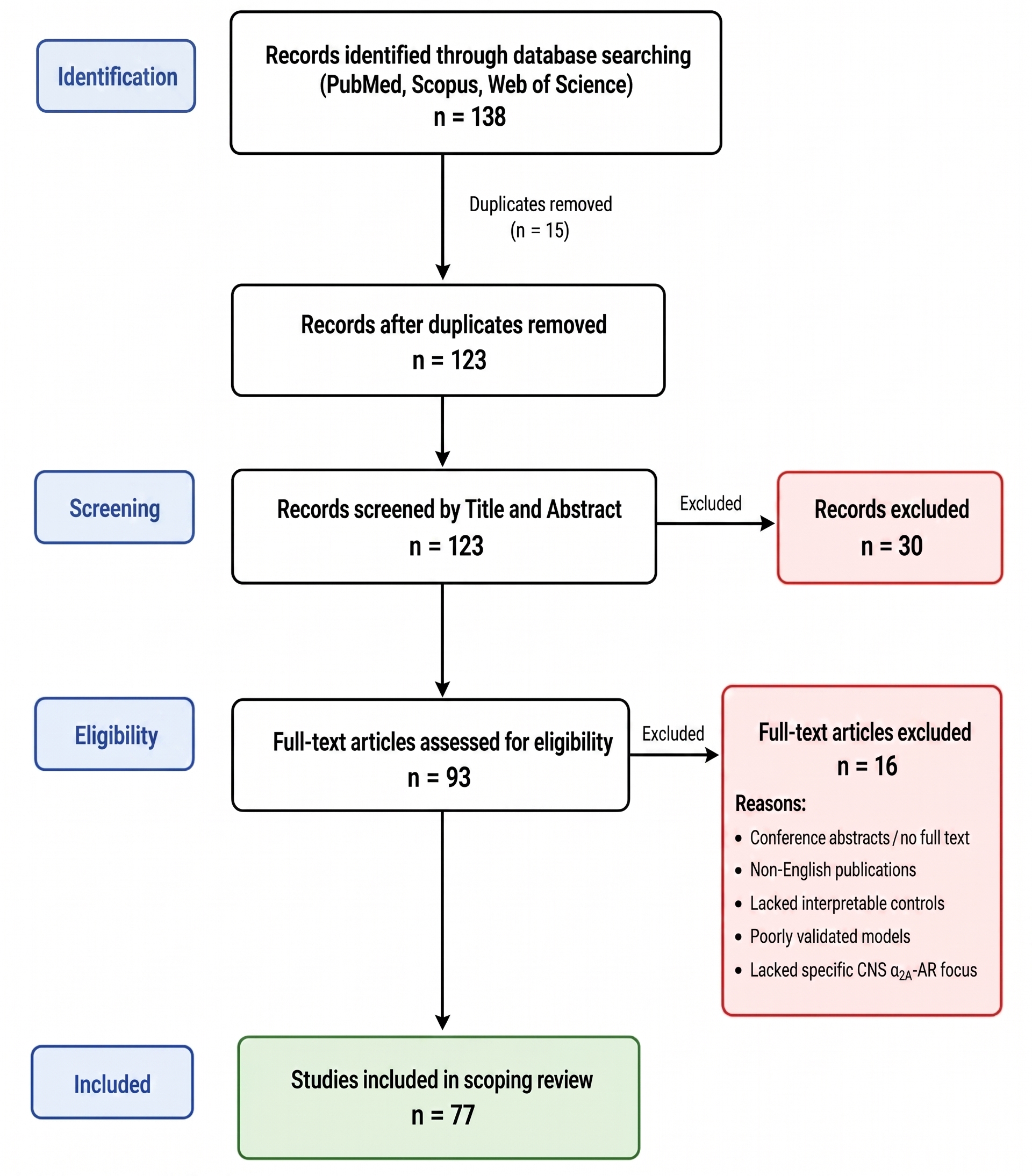
PRISMA-ScR flow diagram of the study selection process. A systematic literature search across PubMed, Scopus, and Web of Science yielded 138 initial records. Following the removal of 15 duplicates, 123 unique records were screened by title and abstract, resulting in the exclusion of 30 irrelevant studies. During the eligibility phase, 93 full-text articles were assessed against the predefined criteria. Of these, 16 were excluded for reasons including lack of full text, language, model validity, or lacking a specific focus on central nervous system α2A-AR biology. Ultimately, 77 preclinical studies met all criteria and were included in the final scoping review.
Characteristics and Principal Findings of Key Preclinical Studies Investigating α2A-AR Modulation
| Study Year | Experimental Model/Preparation | Intervention/Target | Principal Preclinical Findings |
|---|---|---|---|
| Jurgens et al. (2007). | α2A-AR activation | Demonstrated that α2A-AR activation acts directly on recurrent excitatory networks to effectively reduce spontaneous epileptiform burst frequency. | |
| Janumpalli et al. (2008). | Endogenous norepinephrine | Confirmed that the α2A-AR subtype is explicitly required for the antiepileptogenic and seizure-restraining actions of endogenous norepinephrine. | |
| Avoli et al. (2002). | Pharmacological network mapping | Characterized the 4-AP model as a mixed-synaptic state where both excitatory and GABAergic signaling actively drive complex epileptiform synchronization. | |
| Fujita et al. (2013). & Maciulaitiene et al. (2024) | Brimonidine (UK14,304) | Demonstrated robust, direct central neuroprotective properties, promoting neuronal survival, ERK1/2 signaling, and axon growth following acute injury. | |
| Zhang et al. (2021). | α2-adrenergic agonists | Showed that enhancing α2-adrenergic tone significantly mitigates seizure-induced respiratory arrest and prevents fatal cardiorespiratory collapse. | |
| Biggane et al. (2022). | α2A-AR specific ligands | Provided quantitative pharmacological characterization of ligand efficacy, confirming robust suppression of CA3 epileptiform activity via α2A-ARs. | |
| Fink et al. (2023). & Xu et al. (2022) | Biased α2A-agonists (Gi/o preference) | Demonstrated that structure-guided ligands can preferentially activate Gi/o pathways (anticonvulsant/analgesic) while avoiding β-arrestin recruitment (sedation). | |
| Abubakar & Ivanov (2025). | Brimonidine (UK14,304) | Demonstrated state-dependent modulation, showing that α2A-AR agonism preferentially dampens pathological network synchronization while sparing baseline physiological synaptic responses. | |
| Jurgens et al. (2005). | Pharmacological modulation of adrenergic receptors (evaluating the distinct effects of endogenous norepinephrine alongside specific α1-, α2-, and β-adrenergic receptor agonists and antagonists). | Demonstrated that norepinephrine exerts opposing modulatory effects on CA3 network excitability depending on the receptor subtype engaged. Specifically, activation of α2-adrenergic receptors potently suppresses spontaneous epileptiform burst frequency, whereas activation of β-adrenergic receptors increases network burst frequency. This highlights α2-ARs as the primary inhibitory driver of adrenergic seizure suppression in this local circuit. | |
| Ahmed et al. (2001) | Brimonidine (selective α2-adrenergic receptor agonist). | Demonstrated that brimonidine exerts a significant, direct neuroprotective effect on adult rat retinal ganglion cells following ischemic injury induced by elevated intraocular pressure. This highlights the neuroprotective capacity of α2-AR agonism beyond its purely hemodynamic or pressure-lowering effects. | |
| Qu et al. (2019) | Structural and mutational analysis of the α2A-adrenergic receptor (α2A-AR) in complex with a partial agonist and an antagonist to identify specific molecular determinants for ligand binding and G-protein coupling. | Successfully determined the crystal structures of the α2A-AR. The study identified key non-conserved amino acid residues spanning from the ligand-binding pocket (Phe7.39 and Tyr6.55) to the intracellular G-protein coupling region (Ile34.51 and Lys34.56). It demonstrated that these specific residues govern the interplay between partial agonism and biased signaling, providing the critical structural blueprint needed for the rational design of next-generation, highly selective, or pathway-biased α2A-AR therapeutics. | |
| Tavares et al. (1996) | Mapping the neuroanatomical localization and subtype-specific mRNA expression of the α2A- and α2B-adrenergic receptors across distinct brain structures. | Demonstrated that both α2A- and α2B-AR mRNAs are widely distributed throughout the brain. The study found that α2A-AR expression is generally much greater than α2B-AR expression in most central areas (including the cerebral cortex, cerebellum, pons-medulla, and hypothalamus). Notably, this study successfully detected α2A-AR mRNA in the thalamus, the trigeminal nucleus, and both the granule and molecular layers of the cerebellum, highlighting the expansive neuroanatomical footprint of the α2A subtype. | |
| Shen et al. (2020) | Modulation of the central noradrenergic system (investigating endogenous norepinephrine deficiency and evaluating pharmacological interventions aimed at enhancing norepinephrine transmission to prevent SIRA). | Demonstrated that a critical deficiency in central norepinephrine transmission is a primary driver of seizure-induced respiratory arrest (SIRA) in DBA/1 mice. Crucially, the study showed that pharmacologically enhancing noradrenergic tone successfully restored respiratory function and prevented SIRA. This establishes a vital translational link between central adrenergic signaling and the prevention of fatal respiratory collapse, highlighting the neuroprotective potential of targeting these pathways in SUDEP. | |
| Tan et al. (2002) | Pharmacological comparison of full versus partial α2A-adrenergic receptor agonists to evaluate the impact of receptor reserve on therapeutic efficacy and side-effect profiles. | Demonstrated that in a state of reduced receptor reserve, partial α2A-AR agonists can successfully decouple desired therapeutic effects from dose-limiting side effects. Specifically, the study showed that while full agonists caused severe sedation and cardiovascular depression (bradycardia), partial agonists maintained their target therapeutic efficacy with a significantly widened therapeutic window. This provides crucial | |
| Sitnikova et al. (2023) | Pharmacological modulation of α2-adrenergic receptors (evaluating both systemic agonists and antagonists) to determine their specific influence on thalamocortical network excitability and the generation of absence seizures. | Demonstrated a critical, network-specific caveat to α2-AR pharmacology: activation of α2-ARs potently | |
| Szot et al. (1999) | Investigating the fundamental physiological role of the central noradrenergic system by comparing seizure thresholds, propagation, and survival rates between norepinephrine-deficient mice and wild-type controls. | Demonstrated definitively that endogenous norepinephrine serves as a critical, broad-spectrum anticonvulsant mechanism in the brain. The study found that norepinephrine-deficient mice exhibited significantly increased susceptibility to all tested seizure-inducing stimuli, displaying lower seizure thresholds, dramatically more severe seizure phenotypes, and higher seizure-induced mortality compared to wild-type mice. |
αA-Adrenergic Receptor Biology
The Adrenergic System
The adrenergic system is a central regulator of cardiovascular function, metabolism, and arousal 23. Its primary agonists are the catecholamines norepinephrine (NE) and epinephrine, synthesized sequentially from tyrosine in adrenal chromaffin cells and noradrenergic neurons 24. While epinephrine acts primarily as a circulating hormone, NE serves as the principal neurotransmitter in the brain 6. Noradrenergic neurons, located primarily in the locus coeruleus, project widely to the cortex, hippocampus, and amygdala. Following release, NE is cleared via reuptake and metabolism, a process that tightly regulates noradrenergic tone.
Adrenergic Receptors and the αA Subtype
Adrenergic receptors are G protein-coupled receptors (GPCRs) classified into α1, α2, and β families 25. Functionally, these receptors diverge significantly: α1 receptors couple to Gq/11 to increase excitability, whereas β receptors couple to Gs to stimulate cAMP production 26. In contrast, the α2 family (αA, α2B, α2C) couples to Gi/o proteins to exert inhibitory effects 11.
The αA-AR is the predominant subtype in the CNS 27. Mechanistically, agonist binding inhibits adenylyl cyclase via Gαi/o, reducing cAMP/PKA activity. Concurrently, Gβγ subunits inhibit voltage-gated Ca2 channels (VGCCs) and activate G-protein-gated inwardly rectifying K (GIRK) channels 28,29. Through these combined actions, αA-AR activation hyperpolarizes neurons and suppresses neurotransmitter release. Recent structural studies have identified key residues for ligand binding, facilitating the design of biased ligands that may optimize these signaling pathways 12.
CNS Distribution
Immunohistochemical studies reveal a strategic distribution of αA-ARs in seizure-relevant circuits 30. In the brainstem, they function as autoreceptors on the locus coeruleus to regulate NE firing 29. In the forebrain, specifically the hippocampus and neocortex, they are located on presynaptic terminals of Schaffer collaterals and inhibitory interneurons 31. Ultrastructural analysis confirms their localization on presynaptic boutons, supporting their role in gating excitatory transmission 31.
α-2A Adrenoceptor Functions
The molecular identification of different adrenoceptor subtypes spurred research into subtype-selective agents to improve therapeutic effectiveness and reduce off-target effects. Gene targeting studies show that the α₂A adrenoceptor subtype is responsible for many of the classical pharmacological effects linked to non-selective α₂ agonists like clonidine 32. In transgenic mouse models, activating α₂A-adrenoceptors has been associated with hypotension and bradycardia. These responses are diminished or absent when α₂A receptors are genetically disrupted, setting them apart from other α₂ subtypes 32. Additionally, α₂A adrenoceptors play a role in sedation and the central nervous system depressant effects caused by α₂ agonists 32. Neuronal α₂A receptors in the prefrontal cortex are linked to enhanced working memory and cognitive control after selective α₂A activation in primate models 33,34. Although α₂ agonists like clonidine and dexmedetomidine have pain-relieving properties, the involvement of various receptor subtypes and neural pathways makes it difficult to attribute these effects solely to α₂A-adrenoceptors 35.
Pre- and Postsynaptic Mechanisms
Presynaptically, αA-ARs act as "brakes" on synaptic transmission 36. As autoreceptors, they inhibit NE release via negative feedback. As heteroreceptors on glutamatergic terminals, they significantly reduce excitatory postsynaptic potentials (EPSPs) by suppressing presynaptic Ca2+ entry 36. Crucially, this inhibition is more pronounced at excitatory synapses than GABAergic synapses, allowing for a net reduction in network excitability 36. Postsynaptically, αA-AR activation opens GIRK channels, generating an outward K+ current that hyperpolarizes pyramidal neurons and dampens their response to depolarizing inputs 37. Collectively, these receptors act as dynamic gain controllers (Figure 3), regulating the transformation of synaptic inputs into spiking output 38.
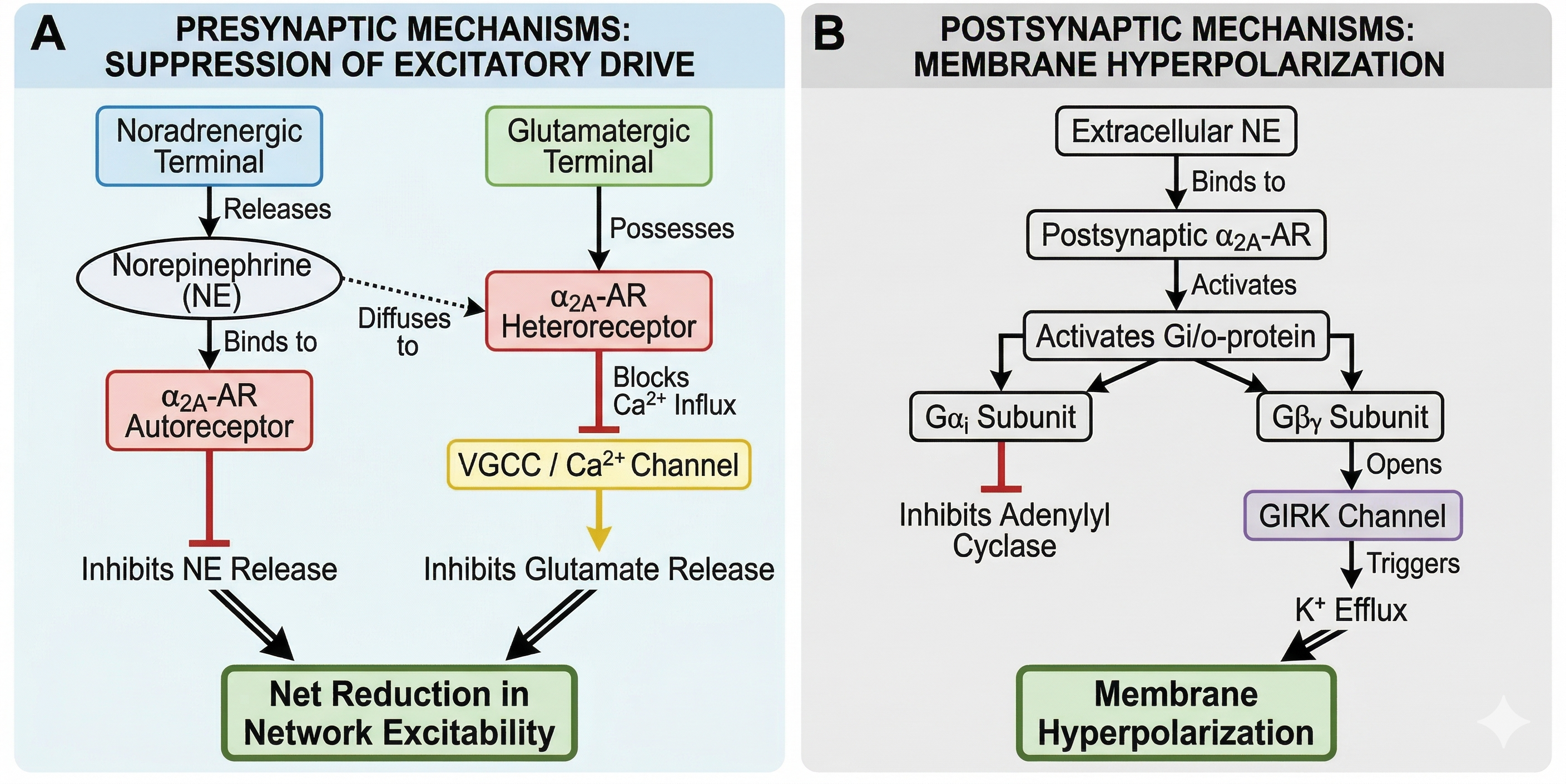
Pre- and Postsynaptic Mechanisms Underlying Synaptic Transmission. This schematic illustrates key presynaptic and postsynaptic processes involved in neurotransmission, including vesicle docking and release at the presynaptic terminal, receptor activation on the postsynaptic membrane, and subsequent ion flux that contributes to synaptic signaling.
The Role of αA-ARs in CNS Excitability and Epilepsy
Glutamatergic and GABAergic Imbalance
Seizures arise when the balance between glutamate-mediated excitation and GABA-mediated inhibition is disrupted 39,40. Under physiological conditions, glutamate activates N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors to drive depolarization, while GABA activates GABA and GABA receptors to hyperpolarize neurons 41. In epilepsy, this homeostasis is lost; reduced GABAergic inhibition or excessive glutamatergic drive leads to runaway excitation and excitotoxicity 42. While current ASMs target these systems directly, their broad modulation often results in cognitive side effects, necessitating more selective neuromodulatory approaches 43.
Noradrenergic Modulation of Epileptiform Activity
The noradrenergic system intersects with these primary signaling pathways to modulate seizure susceptibility 44. Evidence indicates that NE generally exerts anticonvulsant effects; NE-deficient mice exhibit increased seizure susceptibility, which is reversed by restoring NE synthesis 7,44. However, the effect is receptor-dependent: α1 activation can be proconvulsant during stress, and β activation may facilitate excitability 45. Conversely, αA-AR activation consistently suppresses glutamate release and inhibits epileptiform activity 9. This receptor-specific dichotomy suggests that selective αA targeting can harness the anticonvulsant potential of NE while avoiding the proconvulsant actions of other adrenergic subtypes 8,45.
Pharmacology and Actions of Brimonidine (UK14,304)
Clinical Pharmacology and Translational Pharmacokinetics
Brimonidine (UK-14,304) was synthesized as an α2-adrenoceptor-selective imidazoline agonist with reduced lipophilicity compared to clonidine, theoretically to minimize blood-brain barrier (BBB) penetration and consequent sedation 46. However, experimental and clinical data indicate that brimonidine retains pharmacologically significant central nervous system (CNS) access. In pediatric patients, topical ophthalmic administration has precipitated lethargy, unresponsiveness, apnea, bradycardia, and hypotension, confirming its central activity 47,48. Furthermore, animal models demonstrate that radiolabeled brimonidine reaches the optic nerves, optic tracts, and olfactory bulb following ocular dosing, despite negligible systemic blood concentrations. BBB-permeation studies further corroborate brimonidine's measurable capacity to cross into the CNS 49,50. Therefore, brimonidine should be viewed as a ligand with limited but defined brain penetrance, rather than a compound entirely excluded from the CNS.
Importantly, merely increasing BBB penetration would not improve the drug's therapeutic index due to the complex autonomic biology of α2-adrenoceptors. Gene-targeting studies reveal that brainstem αA-receptors mediate the canonical hypotensive response, whereas peripheral α2B-receptors in vascular smooth muscle mediate vasoconstriction, which can counteract central hypotension 51,52. Reflecting this mixed cardiovascular profile, even unilateral topical brimonidine elicits measurable reductions in systemic blood pressure and heart rate in adults 53. This highlights the potent peripheral and systemic effects of the drug, which parallel its established clinical utility in lowering intraocular pressure via specific receptor cascades (Figure 4). Therefore, optimizing this drug class requires achieving CNS target engagement while minimizing both central sympatholysis and peripheral vascular receptor occupancy.
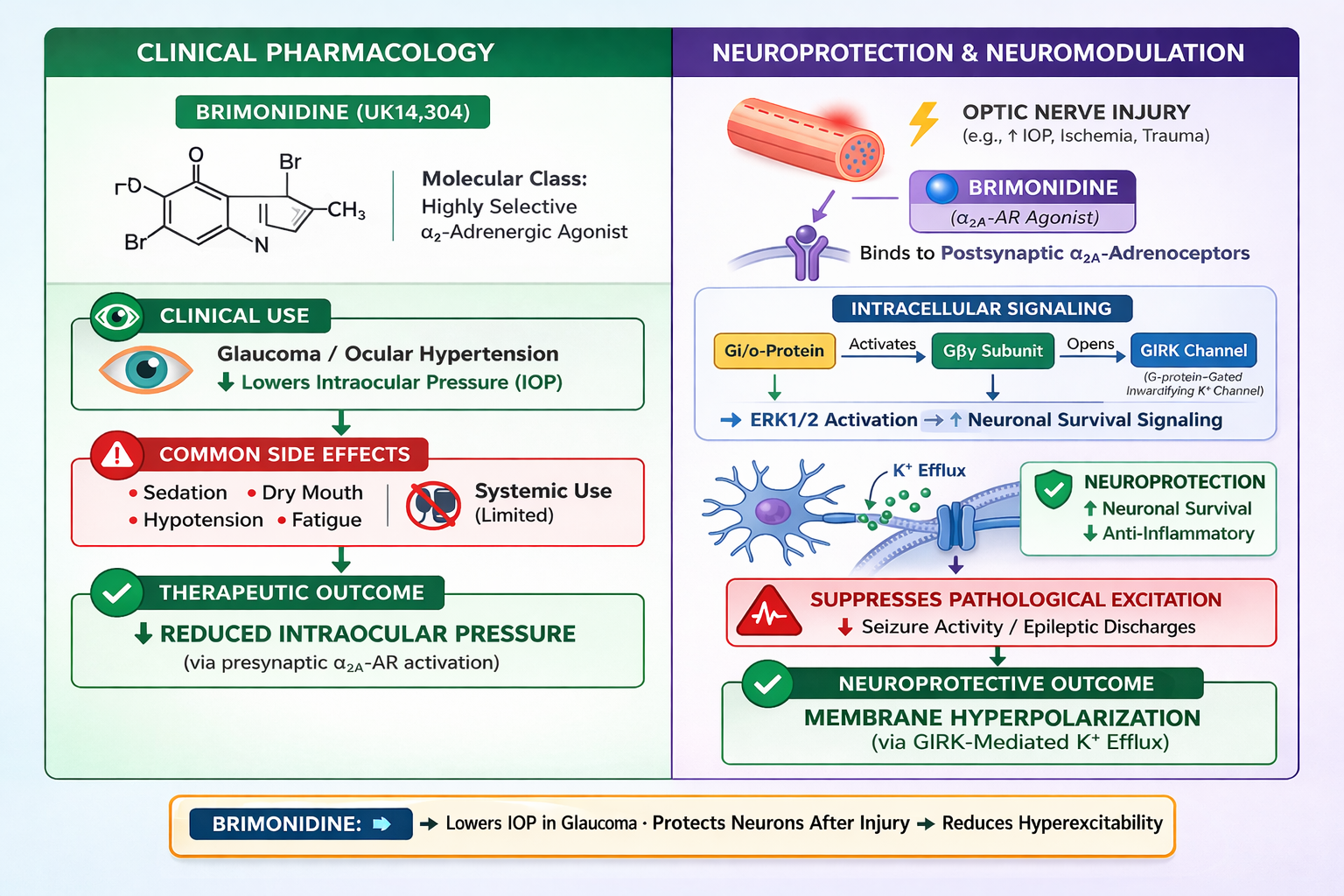
Pharmacological Profile and Physiological Actions of the Selective α2-adrenoceptor Agonist brimonidine (UK-14,304). Brimonidine is a potent and selective α2-adrenoceptor agonist utilized both as a pharmacological reference tool and a therapeutic agent. Agonist binding to Gi/o-coupled α2-adrenoceptors activates signaling cascades that suppress neuronal excitability and neurotransmitter release. This is mediated primarily through Gαi-dependent inhibition of adenylyl cyclase and cAMP production, alongside Gβγ-dependent inhibition of voltage-gated Ca2+ channels and activation of G protein-coupled inwardly-rectifying K+ (GIRK) channels. Clinically, brimonidine is indicated for lowering intraocular pressure (IOP). This therapeutic effect is attributed to a dual mechanism involving suppressed aqueous humor production and increased uveoscleral outflow.
Consequently, future brimonidine-inspired ligands must be optimized across receptor subtype, signaling bias, and pharmacokinetic exposure dimensions. First, candidate molecules should exhibit selectivity for the αA-subtype over α2B in both binding affinity and intrinsic efficacy. Brimonidine's functional α2-selectivity is driven largely by this efficacy differential rather than an absolute affinity gap. Second, next-generation ligands must circumvent the broad, high-efficacy signaling profile of canonical imidazoline agonists. Brimonidine and dexmedetomidine function as robust agonists for both G protein activation and β-arrestin-2 recruitment at the human αA-receptor. Conversely, recently developed structure-guided αA-agonists exhibit preferential Gi/o signaling with negligible β-arrestin recruitment, preserving in vivo analgesia without inducing sedation 53. While it remains to be definitively proven that biased signaling eliminates hypotension, this establishes a clear medicinal chemistry directive: preserve orthosteric interactions required for αA activation while avoiding conformations linked to broad transducer engagement. Recent cryo-electron microscopy (cryo-EM) structures of the αA-receptor offer a template for this targeted design, highlighting conserved binding-pocket residues such as D128, Y431, and F42754.
Third, pharmacokinetic optimization must prioritize unbound brain exposure over crude lipophilicity. In CNS drug discovery, the unbound brain-to-plasma partition coefficient is a superior metric to nominal BBB permeability, as it accounts for the net effects of influx, efflux, and non-specific tissue binding 55. An optimal brimonidine successor requires sufficient unbound CNS exposure to engage αA-dependent neuroprotective or spinal circuits, alongside low unbound plasma concentrations to limit systemic α2-receptor occupancy 56. Furthermore, emerging research suggests that mitigating cardiovascular liability may require circuit-level modulation rather than exclusively intensifying αA agonism. For instance, ADRIANA, a recently identified α2B-selective antagonist, elevates spinal noradrenaline and induces αA-dependent analgesia without cardiovascular side effects in murine and non-human primate models 57. This underscores a critical design principle: future CNS-active therapeutics must integrate robust brain exposure with αA-selective efficacy and peripheral α2B sparing, rather than pursuing global α2-adrenoceptor agonism.
Neuroprotection and Neuromodulation
Beyond its ocular effects, brimonidine exhibits neuroprotective properties 58. In optic nerve injury models, it promotes neuronal survival via ERK1/2 signaling and suppression of inflammatory pathways 59. In the context of epilepsy, the drug's ability to penetrate neural tissue and robustly activate Gi/o signaling makes it an attractive candidate for suppressing pathological excitation 60.
Preclinical Evidence in Epileptic Models
Hippocampal Slice Models
Ex vivo hippocampal slice studies confirm the anticonvulsant properties of brimonidine, but understanding its true mechanism requires comparing distinct epileptogenic models. In early CA3 models, epileptiform bursting was induced by pharmacologically impairing GABAergic inhibition. Under these disinhibited conditions, αA-adrenergic receptor (αA-AR) activation successfully reduced spontaneous burst frequency. This demonstrates that αA-ARs can suppress hyperexcitability directly by attenuating the recurrent excitatory CA3 network, rather than merely restoring lost GABAergic tone 9,38. However, the pharmacological landscape shifts significantly when epileptiform activity is induced by 4-aminopyridine (4-AP). Unlike standard disinhibition models, 4-AP blocks voltage-gated potassium channels, which broadly enhances neurotransmitter release from both excitatory and inhibitory terminals. Consequently, the 4-AP model preserves and actively recruits GABAergic signaling into the epileptiform pattern. In hippocampal slices, 4-AP generates complex synchronous activity, ranging from fast CA3-driven glutamatergic interictal events to slower GABA-dependent discharges 61. Evaluating a drug in this environment tests its efficacy in a circuit where excitation, inhibition, and propagation dynamics remain intact and highly complex.
This model-specific context clarifies recent findings regarding brimonidine's efficacy. In adult mouse hippocampal slices treated with 4-AP, brimonidine significantly increased the interval between interictal-like discharges while leaving physiological synaptic responses largely unchanged 62. This targeted response reflects state-dependent modulation. In the older CA3 model, αA-AR signaling is tested against a simplified substrate where recurrent excitation dominates. In the 4-AP model, the same receptor system is tested in a mechanistically mixed epileptiform state, where preserved inhibitory signaling reshapes the drug's apparent effect. Ultimately, comparing these ex vivo studies indicates that brimonidine is not acting as a uniform central nervous system depressant. Its efficacy heavily depends on the pathological state of the network. When challenged by the robust, mixed-synaptic hyperactivity of the 4-AP model, αA-AR agonism preferentially dampens pathological network synchronization while sparing baseline physiological transmission 9,61,62. This state-dependency provides a vital theoretical framework for how αA-agonists might control focal seizures without indiscriminately suppressing normal hippocampal function.
SUDEP and Mortality Models
Beyond seizure suppression, the noradrenergic system is critical for preventing Sudden Unexpected Death in Epilepsy (SUDEP) 63. In DBA/1 mice, a well-established model of SUDEP, deficiencies in norepinephrine synthesis correlate with seizure-induced respiratory arrest 63. Pharmacological interventions that enhance noradrenergic tone or activate α2-adrenergic receptors have been shown to reduce the incidence of respiratory arrest significantly 14. While clonidine, a non-selective α2 agonist, was utilized in these studies, the high density of αA receptors in brainstem respiratory centers suggests this subtype may play a predominant role in maintaining respiratory drive during ictal events 14. Thus, α2-adrenergic modulation offers a potential dual therapeutic benefit: mitigating cortical hyperexcitability and preventing fatal cardiorespiratory collapse.
Targeted Delivery
To circumvent the systemic side effects of α2 agonists (e.g., hypotension), novel delivery systems are under investigation 13. Approaches such as electrophoretic drug delivery allow for focal, on-demand administration of ASMs directly to the epileptogenic zone 13. Applying such technologies to brimonidine could maximize local antiseizure efficacy while minimizing peripheral exposure (Proctor et al., 2018).
Therapeutic Implications and Future Directions
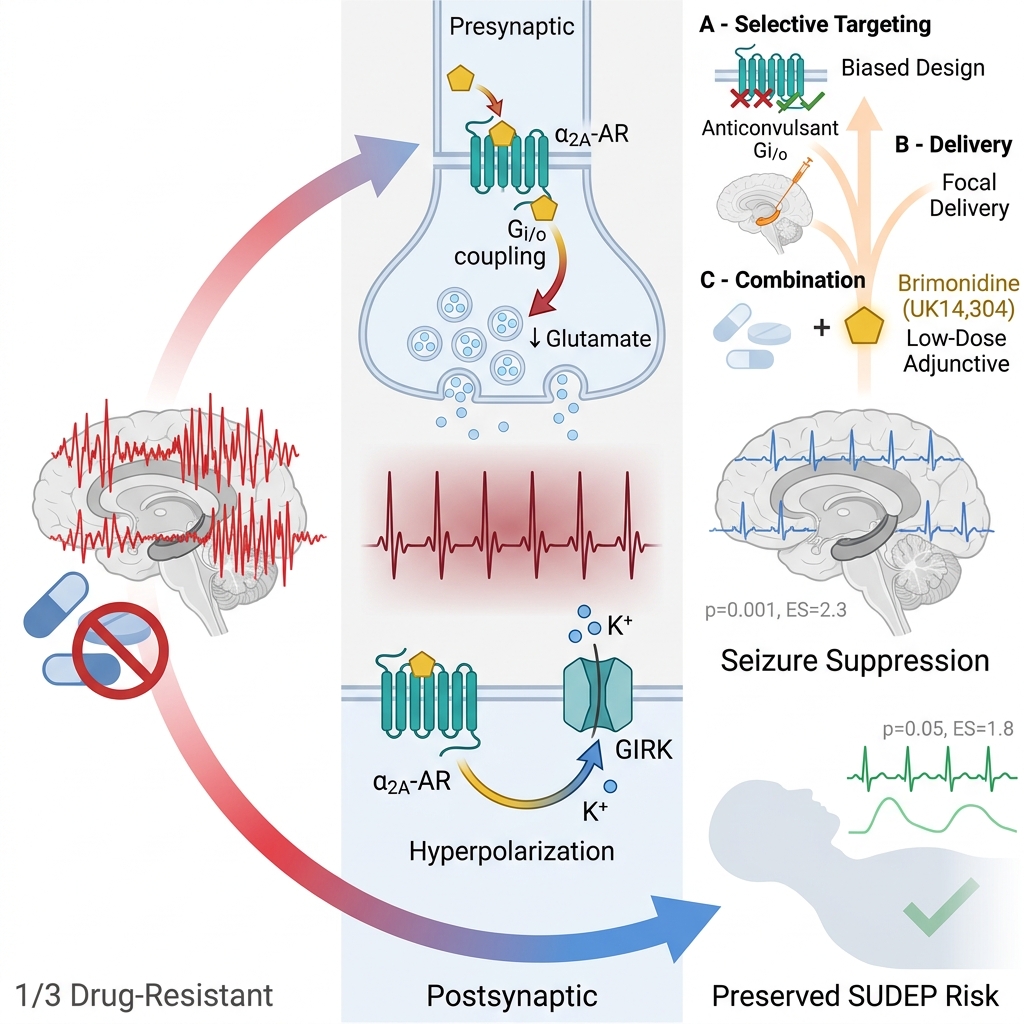
The evidence reviewed supports αA-ARs as high-value targets for next-generation epilepsy therapies (Löscher et al., 2013). To translate this potential into clinical practice, several strategic avenues must be pursued (Figure 5):
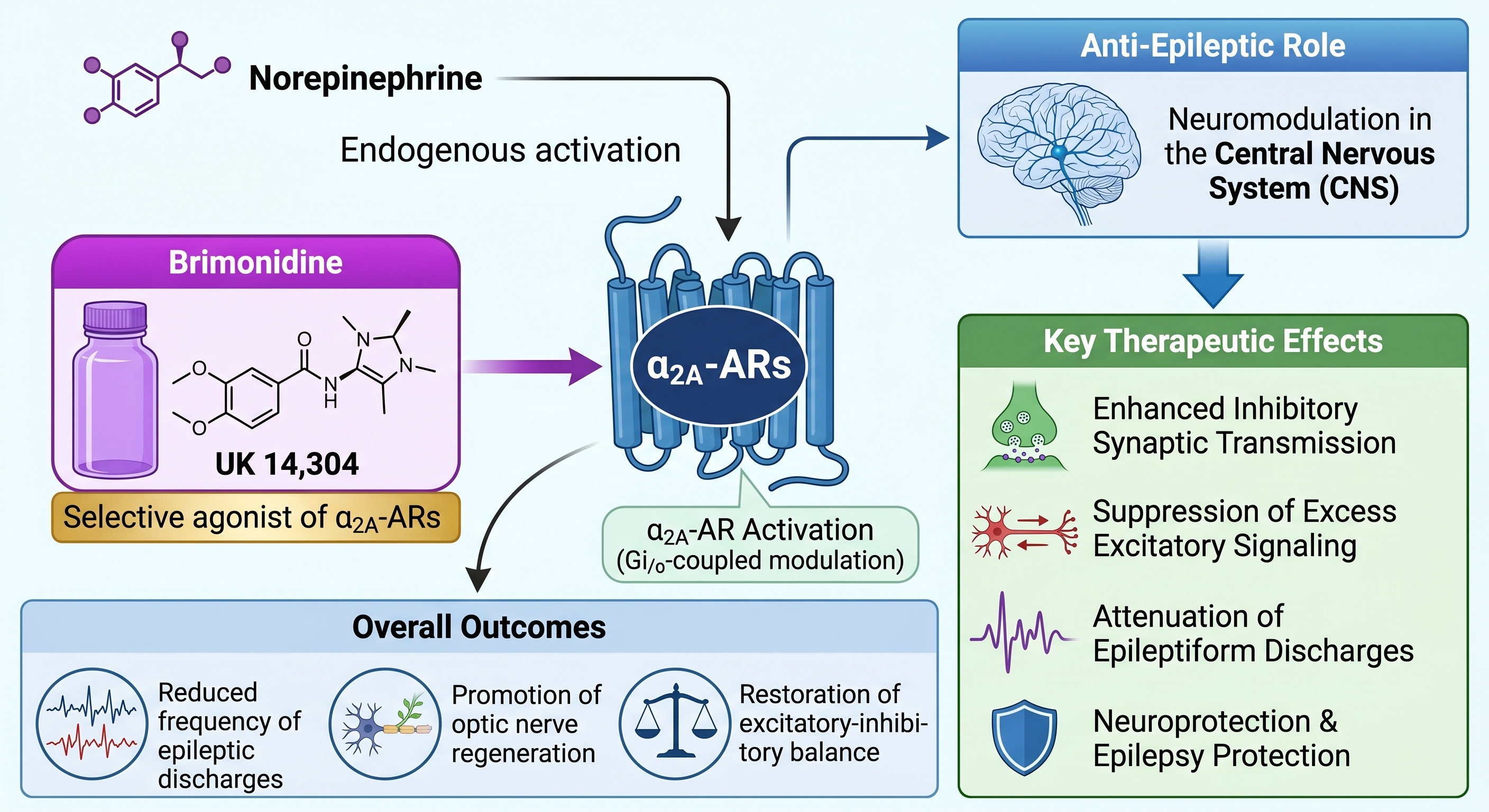
α₂A-Adrenergic Receptor-Mediated Neuromodulation and Its Anti-Epileptic Effects. This schematic illustrates the neuromodulatory pathways initiated by the activation of central α₂A-adrenergic receptors (α₂A-ARs). Both endogenous norepinephrine and the selective pharmacological agonist brimonidine (UK14,304) drive α₂A-AR activation via Gi/o-coupled modulation. This targeted activation results in Central Nervous System (CNS) neuromodulation that serves an anti-epileptic role by driving key therapeutic effects: enhanced inhibitory synaptic transmission, suppression of excess excitatory signaling, attenuation of epileptiform discharges, and neuroprotection and epilepsy protection. Ultimately, these targeted mechanistic pathways yield overall outcomes that include a reduced frequency of epileptic discharges, the promotion of optic nerve regeneration, and the restoration of excitatory-inhibitory balance.
Subtype-Selective and Biased Ligands
Leveraging structural biology to design "biased" agonists that favor G-protein anticonvulsant pathways over β-arrestin pathways linked to sedation 12. Recent advances in G protein-coupled receptor (GPCR) structural biology have fundamentally transformed the approach to targeting the αA-adrenergic receptor. Traditional agonists, such as clonidine and brimonidine, act as "balanced" ligands; they uniformly recruit both Gi/o protein signaling pathways (which mediate the desired presynaptic anticonvulsant effects) and β-arrestin-2 pathways (which are heavily implicated in dose-limiting adverse events, such as profound sedation and cardiovascular depression). The proposal that future therapeutic success lies in "biased" agonism is rooted in the pharmacological ability to decouple these distinct intracellular cascades. Current evidence indicates that αA-adrenoceptor activation is the subtype most directly linked to anticonvulsant effects, as αA signaling suppresses epileptiform activity and contributes to endogenous seizure restraint 9,64,65. However, effective seizure control does not require indiscriminate activation of all downstream pathways. Studies in mice show that sedative responses to α2 agonists are influenced by arrestin-associated signaling machinery, whereas partial αA activation can preserve selected physiological effects without proportionate sedation 66,67.
By leveraging high-resolution cryo-electron microscopy (cryo-EM) structures of the human αA-receptor in its active state, drug discovery has moved from empirical screening to rational, structure-guided design (Xu et al., 2022). For example, recent large-scale computational docking campaigns have successfully identified novel, non-imidazoline αA-agonists that exhibit profound signaling bias. These molecules preferentially trigger Gi/o signaling with virtually undetectable β-arrestin recruitment 68.
At the molecular level, this functional selectivity is achieved by engineering ligands that selectively interact with specific conserved residues within the orthosteric binding pocket—most notably D128, Y431, and F42712,68. By finely tuning the steric and electrostatic contacts with these specific amino acids, medicinal chemists can stabilize a distinct receptor conformation. This precise conformation permits the intracellular loops to engage G proteins but sterically hinders the intracellular structural rearrangements required for β-arrestin coupling. In preclinical in vivo models, these structure-derived biased ligands successfully preserved centrally mediated analgesia without inducing sedation 68. Applying this structural paradigm to epilepsy represents a highly promising translational frontier. Engineering biased αA-agonists could theoretically isolate the state-dependent suppression of epileptiform discharges from the systemic sympatholytic and sedative burdens that have historically precluded the chronic systemic use of these drugs in clinical neurology.
Focal Delivery Systems
Utilizing intracranial catheters or implantable devices to deliver αA agonists specifically to seizure foci, beneficial for drug-resistant focal epilepsies 13. Systemic administration of αA-agonists is heavily restricted by dose-limiting peripheral cardiovascular effects and generalized sedation. To circumvent these translational barriers, future strategies must leverage focal delivery systems to treat drug-resistant focal epilepsies. This approach utilizes intracranial catheters or implantable micro-infusion pumps to deliver αA-agonists directly into the precisely mapped epileptogenic zone. By bypassing the blood-brain barrier and systemic circulation, focal delivery achieves high, sustained therapeutic drug concentrations exactly where pathological hypersynchrony originates 69. Furthermore, advancing these implantable devices could allow for integration with responsive neurostimulation (RNS) technology, creating a closed-loop system that triggers micro-infusions of the drug only upon the detection of pre-seizure electrophysiological biomarkers 70. Ultimately, restricting αA-AR activation exclusively to the seizure focus maximizes the local anticonvulsant efficacy while entirely eliminating the peripheral sympatholytic and broad cognitive side effects that have historically limited this drug class.
Adjunctive Therapy
Employing low-dose αA agonists alongside standard ASMs to enhance seizure control and reduce SUDEP risk without compounding side effects 14. Monotherapy with standard antiseizure medications (ASMs) frequently fails to achieve complete seizure freedom or requires high doses that cause intolerable cognitive impairment. Employing low-dose αA-agonists as an adjunctive therapy offers a highly synergistic pharmacological approach. Because standard ASMs predominantly target postsynaptic ion channels or GABAergic pathways, the addition of a presynaptic αA-agonist provides a complementary mechanism to further attenuate pathological glutamate release. Importantly, utilizing this drug class at a low dose enhances seizure control while avoiding the profound sedation and hypotension that typically preclude high-dose α2-agonist monotherapy. Furthermore, this adjunctive strategy may actively mitigate the risk of Sudden Unexpected Death in Epilepsy (SUDEP). SUDEP is fundamentally driven by massive, seizure-induced autonomic dysregulation, including severe post-ictal sympathetic storms and fatal cardiac arrhythmias 71. Because αA-receptors function as central sympatholytics, their targeted activation can dampen these catastrophic neurocardiac reflexes, potentially offering a dual-action therapy that reduces both seizure frequency and the likelihood of fatal autonomic collapse 72.
Disease Modification
Investigating whether the neuroprotective pathways activated by UK14,304 can retard epileptogenesis or prevent network reorganization 58. Current antiseizure medications are overwhelmingly symptomatic; they suppress seizures but do not alter the underlying pathological progression of the disease. A critical frontier in epilepsy research is determining whether the neuroprotective properties of αA-agonists, such as brimonidine, can offer true disease modification. Epileptogenesis—the process by which a normal brain develops chronic epilepsy following an initial insult like status epilepticus or trauma—is fundamentally driven by acute glutamate excitotoxicity, neuroinflammation, and subsequent aberrant network reorganization, including mossy fiber sprouting and neuronal death 73. Because αA-agonists potently inhibit presynaptic glutamate release, they directly mitigate the primary driver of excitotoxic cell damage. Furthermore, preclinical studies across various neurodegeneration and brain injury models demonstrate that α2-adrenoceptor activation upregulates neurotrophic factors and activates anti-apoptotic cellular cascades 74,75. By combining this robust anti-excitotoxic action with direct cellular neuroprotection, future translational research must investigate whether early intervention with αA-agonists can successfully arrest epileptogenesis and prevent pathological network rewiring, ultimately altering the natural history of the disease rather than merely masking its symptoms.
Clinical Translation and Biomarker-Driven Trials
Bridging the translational gap between rodent ex vivo models and human focal epilepsy requires rigorous, proof-of-concept clinical trial designs. Currently, clinical evidence for αA-mediated neuroprotection or seizure suppression in the human central nervous system remains limited and inconclusive 18,19. To overcome this barrier, future early-phase trials must integrate robust, quantifiable biomarkers rather than relying solely on patient-reported seizure diaries. Non-invasive tools, such as high-density electroencephalography (hdEEG), should be utilized to quantify targeted reductions in interictal epileptiform discharges following acute drug administration. Furthermore, neuroimaging techniques like receptor-specific positron emission tomography (PET) can be deployed to confirm central target engagement and verify that the drug reaches the necessary brain structures at therapeutic concentrations.
In addition to non-invasive biomarkers, translational neurology must leverage specialized clinical environments. "Window of opportunity" (or Phase 0) trials in the epilepsy monitoring unit represent an ideal framework for testing novel αA-agonists 76. In these studies, patients with drug-resistant focal epilepsy who are undergoing invasive intracranial EEG (iEEG) monitoring are evaluated to assess the acute effects of the drug. This setup allows clinicians to directly and precisely measure the drug's local efficacy in suppressing pathological bursting within the exact human epileptogenic zone, overcoming the well-documented limitations and inaccuracies of patient-reported seizure diaries 77. Additionally, if the patient proceeds to surgical resection, the removed brain tissue can undergo molecular analysis to verify αA-AR pathway activation. By combining precise iEEG electrophysiological readouts with direct molecular validation, these trial designs can firmly establish the therapeutic window of αA-AR modulation before advancing to large-scale, chronic systemic trials 73,76.
Conclusion
In summary, the α-2A adrenergic receptor represents a mechanistically distinct target for seizure control, offering a way to modulate the "gain" of neural circuits. Brimonidine (UK14,304) serves as a powerful proof-of-concept molecule, demonstrating that selective αA activation can inhibit pathological discharges while sparing physiological transmission. Future translational success will depend on optimizing delivery methods and developing biased ligands to dissociate the potent antiseizure effects from systemic cardiovascular risks. If achieved, αA-AR modulation could significantly expand the therapeutic toolkit for drug-resistant epilepsy.
Abbreviations
αA-AR: αA-adrenergic receptor; ADRA2A: Adrenoceptor Alpha 2A gene; AMPA: alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; ASM: antiseizure medication; BBB: blood-brain barrier; Ca: calcium ion; cAMP: cyclic adenosine monophosphate; CNS: central nervous system; Cryo-EM: cryo-electron microscopy; EPSP: excitatory postsynaptic potential; GABA: gamma-aminobutyric acid; GIRK: G protein-coupled inwardly rectifying potassium channel; GPCR: G protein-coupled receptor; hdEEG: high-density electroencephalography; iEEG: intracranial electroencephalography; IOP: intraocular pressure; K: potassium ion; NE: norepinephrine; NMDA: N-methyl-D-aspartate; PCC: Population–Concept–Context; PET: positron emission tomography; PRISMA-ScR: Preferred Reporting Items for Systematic Reviews and Meta-Analyses extension for Scoping Reviews; RNS: responsive neurostimulation; SIRA: seizure-induced respiratory arrest; SUDEP: Sudden Unexpected Death in Epilepsy; SWDs: spike-wave discharges; VGCC: voltage-gated calcium channel.
Acknowledgments
The authors would like to acknowledge the support and efforts of the Petroleum Technology Development Fund (PTDF), Nigeria, for their sponsorship.
Author’s contributions
All equally contributed to this work, read and approved the final manuscript.
Funding
None.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have not used generative AI (a type of artificial intelligence technology that can produce various types of content including text, imagery, audio and synthetic data).
Competing interests
The authors declare that they have no competing interests.