

Genetic link between Fetuin-A and Type 2 Diabetes Mellitus: A Systematic Narrative Review
- Department of Biochemistry, Integral Institute of Medical Sciences and Research (IIMSR), Integral University, Lucknow, Uttar Pradesh, India
- Department of General Medicine, Integral Institute of Medical Sciences and Research (IIMSR), Integral University, Lucknow, Uttar Pradesh, India
- Deputy Medical Superintendent, Integral Institute of Medical Sciences and Research (IIMSR), Integral University, Lucknow, Uttar Pradesh, India
- Department of Basic Medical Science, Integral Institute of Allied Health Sciences & Research (IIAHSR), Integral University, Lucknow, Uttar Pradesh, India
Abstract
Introduction: The multifunctional glycoprotein Fetuin-A, encoded by the alpha-2-Heremans-Schmid glycoprotein (AHSG) gene, is highly implicated in insulin resistance and Type 2 Diabetes Mellitus (T2DM). As a hepatokine, Fetuin-A regulates systemic inflammation, lipid metabolism, and insulin receptor signaling. Genetic variants of the AHSG gene alter plasma Fetuin-A concentrations, thereby modulating an individual's susceptibility to metabolic syndrome and T2DM. This review provides an in-depth analysis of the genetic link between circulating Fetuin-A levels and T2DM risk, with a specific focus on the Indian population.
Methodology: The Scopus, PubMed, and Web of Science databases, alongside the Google Scholar search engine, were queried to retrieve relevant literature. Search terms included: “Fetuin-A and T2DM,” “Fetuin-A gene variants and T2DM,” “Fetuin-A gene variants and circulating Fetuin-A,” “circulating Fetuin-A and T2DM,” “AHSG gene variants and T2DM,” and “genetic link between Fetuin-A and T2DM in Indian Population.” The review included genome-wide association studies (GWAS), original research, case-control studies, and case-cohort studies. Eligibility was restricted to full-text articles published in English up to the end of February 2026.
Results: A total of 493 articles were initially retrieved; of these, 13 met the inclusion criteria for this systematic narrative review. The analysis revealed that the most frequently studied AHSG variants—rs4917, rs4918, rs248, rs256, rs2518136, rs2248690, rs6809265, rs2070633, and rs2070635—have been evaluated across diverse ethnic populations.
Conclusion: The AHSG Thr256Ser (rs4918) single nucleotide polymorphism (SNP) is associated with T2DM in South Indian populations. Specifically, the mutant GG genotype of rs4918 appears to correlate with reduced circulating Fetuin-A levels. Conversely, elevated systemic Fetuin-A is strongly associated with insulin resistance and incident T2DM, suggesting that circulating Fetuin-A may serve as a viable predictive biomarker for the development of diabetes.
Introduction
Type 2 diabetes mellitus (T2DM) is a complex, multifactorial metabolic disorder arising from intricate interactions among genetic, environmental, and behavioral factors. A fundamental hallmark of T2DM is insulin resistance, a state in which peripheral tissues—predominantly skeletal muscle, the liver, and adipose tissue—exhibit an impaired response to insulin 1. This defect diminishes cellular glucose uptake, culminating in chronic hyperglycemia. Over time, persistent hyperglycemia can lead to severe microvascular and macrovascular complications, including cardiovascular disease (CVD), nephropathy, retinopathy, and neuropathy 2.
Extensive genome-wide association studies (GWAS) have identified numerous susceptibility loci, underscoring the significant genetic component of T2DM 3. Despite these advancements, the precise genetic and molecular underpinnings of the disease remain incompletely elucidated. Beyond established mechanisms such as pancreatic beta-cell dysfunction and insulin receptor mutations, the pathophysiological role of hepatokines—liver-derived secretory proteins that exert systemic metabolic effects—is garnering increased attention. Hepatokines have emerged as critical mediators in the pathogenesis of insulin resistance and related metabolic disorders 4.
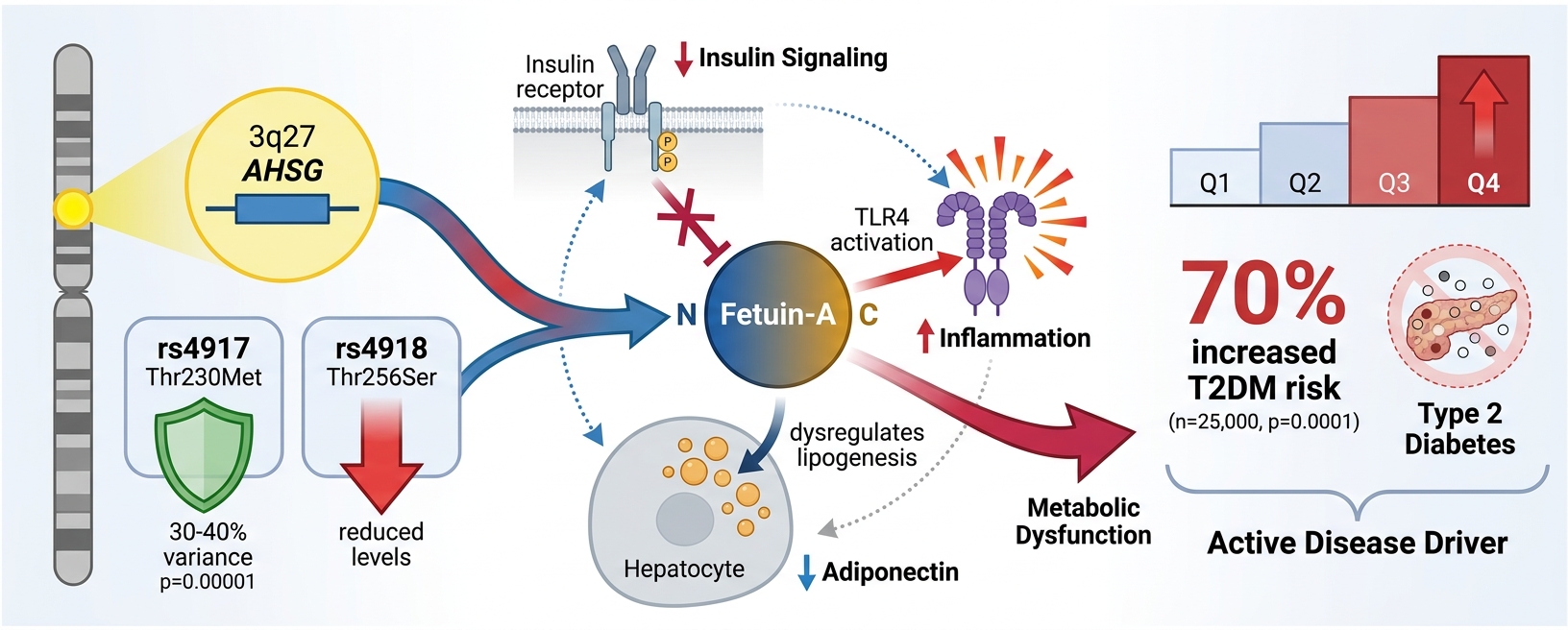
One hepatokine of particular recent interest is Fetuin-A, encoded by the alpha-2-Heremans-Schmid glycoprotein (AHSG) gene located on chromosome 3q27. Notably, a genome-wide linkage study identified chromosome 3q27 as a novel and highly susceptible locus for diabetes 5. The AHSG locus has been previously linked to insulin resistance and components of metabolic syndrome (MetS), with genetic variants associating with altered circulating levels of Fetuin-A 6. Initially characterized as a potent inhibitor of ectopic calcification in mineral metabolism, Fetuin-A has since moved to the forefront of metabolic research due to its newly elucidated roles in insulin resistance, inflammation, and lipid metabolism.
This review aims to investigate the genetic association between Fetuin-A and T2DM specifically within the Indian population. Furthermore, we will examine the pathophysiological mechanisms through which Fetuin-A contributes to the development of T2DM, with a particular focus on its functions in disrupting lipid metabolism, modulating inflammation, and attenuating insulin signaling.
Methodology
A comprehensive literature search was conducted across the Scopus, PubMed, and Web of Science databases, supplemented by the Google Scholar search engine. Articles were retrieved using the following keywords and search phrases: “Fetuin-A and T2DM”, “Fetuin-A gene variants and T2DM”, “Fetuin-A gene variants and Circulatory Fetuin-A”, “Circulatory Fetuin-A and T2DM”, “AHSG gene variants and T2DM”, and “Genetic link between Fetuin-A and T2DM in Indian Population”.
The eligibility criteria for inclusion in this review were defined as follows: original research articles—specifically encompassing genome-wide association studies (GWAS), case-control studies, and case-cohort studies. Furthermore, studies were required to be available as full-text articles published in the English language up to the end of February 2026. The detailed article retrieval and selection process is illustrated in the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram (Figure 1).
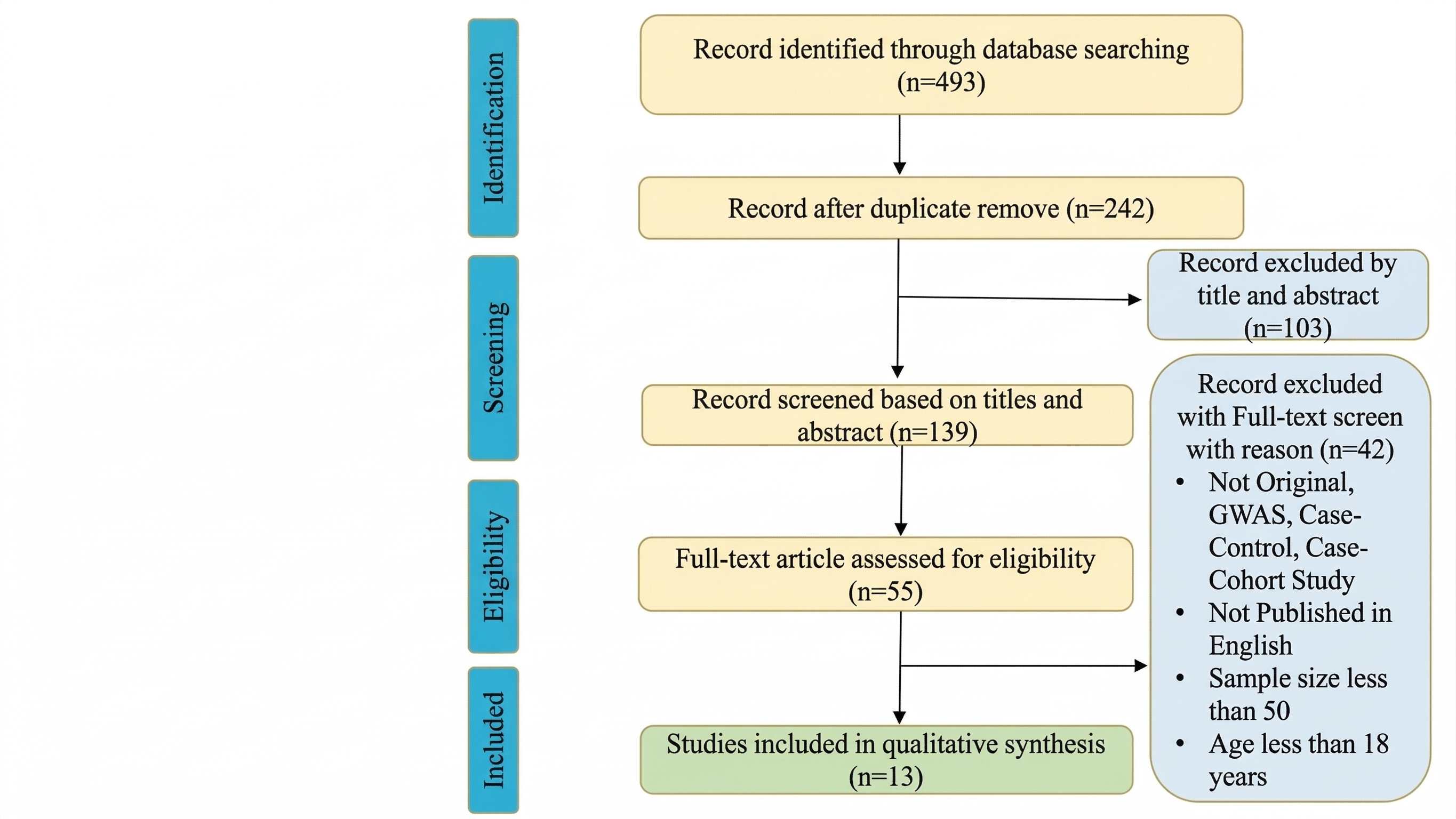
Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram illustrating the detailed article retrieval and selection process. The diagram outlines the progression from the initial identification of 493 articles down to the final 13 articles that met the inclusion criteria for this systematic narrative review.
Results
A total of 493 articles were retrieved. Out of this, only 13 articles were found suitable to include in this systematic narrative review. The most studied AHSG gene variants (encoding Fetuin-A) rs4917, rs4918, rs248, rs256, rs2518136, rs2248690, rs6809265, rs2070633, and rs2070635 were found in different ethnic populations, summarized in Table 1
Summary of Genetic Link between Fetuin-A and T2DM. This table summarizes the most studied
| Author (Year) | Study Design | Population & Sample size (n) | Gene Variants / Circulatory Fetuin-A | Methods | Conclusion |
|---|---|---|---|---|---|
| Andersen G et al (2008) | Case-control | White Danish Population (n=7,683) | 07 AHSG SNPs | Taqman allelic discrimination or chip-based MALDI-TOF Mass Spectrometry | rs2518136 polymorphisms were associated with type 2 diabetes (p<0.006) |
| Jensen MK et al (2013) | Community-based Follow-up | Caucasian (n=2893) and African American (n=542) (T2DM n=259) |
Common Circulatory Fetuin-A | Genotyping Array and ELISA Method | Common |
| Jensen MK et al (2017) | GWAS | European (n=9055) and African Americans (n=2119) | 34 AHSG SNPs | GWAS Meta-Analysis |
rs4917 a known coding SNP in exon 6 that is associated with a 0.06 g/l (∼13%) lower fetuin-A level. This variant alone explained 14% of the variation in fetuin-A levels. |
| Kröger J et al (2018) | GWAS | European Population (n=10020) | 05 AHSG SNPs |
GWAS Mendelian Randomization Analysis Immunoturbidimetric Method |
Fetuin-A showed significant impact on insulin sensitivity and secretion. This study does not support the relationship between Fetuin-A and risk of T2DM in general population. |
| Wang Y et al (2019) | Case-control |
Chinese Population (n=1116) | Circulatory Fetuin-A | ELISA Method | Raised fetuin-A levels were positively associated with T2DM risk in this Chinese population even after adjustment for BMI and other lifestyle risk factors. |
| Morsy EY et al (2020) | Case-control |
Egyptian Population (n=120) |
AHSG SNPs rs248 and rs256 | PCR–RFLP genotyping |
Increased serum Fetuin-A is associated with insulin resistance and increased risk of atherosclerosis in patients with T2DM. TT (rs248) and GG (rs256) polymorphisms may be associated with lower risk of DKD. |
| Susairaj P et al (2021) | Case-control |
Asian Indian (n=144) | Circulatory Fetuin-A | ELISA Method | Elevated Fetuin-A levels are strongly associated with incident T2DM. It may act as a predictive biomarker for development of diabetes. |
| Umapathy D. et al. (2022) | Case-control |
South-Indian (n = 975 ) | AHSG Thr256Ser (rs4918 / C>G) |
PCR–RFLP genotyping ELISA Method |
Mutant GG Genotype may be responsible to reduce the circulatory Fetuin-A levels. |
| Birukov A. et al (2022) | Prospective cohort study |
EPIC-Potsdam / other population cohorts (European ancestry). (n=587) | Circulatory Fetuin-A | ELISA Method | Fetuin-A was linearly inversely associated with microvascular complications in T2DM patients. |
| Pathak AK et al (2023) | Case-control |
North Indian (n=314) | Circulatory Fetuin-A | ELISA Method | Fetuin-A levels were found 3-times higher in T2DM patients than in non-T2DM ( |
| Ali L et al (2023) | GWAS |
UK Biobank (n= ∼412,444) |
AHSG SNPs rs11017848 | GWAS Meta-Analysis | Genetically predicted etuin-A is associated with T2DM. |
| Gupta R et al (2024) | Case-control |
South Indian (n=648) | Circulatory Fetuin-A | ELISA Method | Fetuin-A has not showed any significant association with T2DM patients. |
| Govril et al (2025) | Observational study |
Romania (n=51) | Circulatory Fetuin-A | ELISA Method | Fetuin-A appears to be linked to lipid abnormalities in T2DM and contributed to CVD. |
Fetuin-A: Structure and Function
Fetuin-A, also known as alpha-2-Heremans-Schmid glycoprotein (AHSG), is a multifunctional glycoprotein primarily synthesized and secreted by hepatocytes. In healthy individuals, it circulates in the plasma at relatively high concentrations (0.4–0.8 g/L). The pre-Fetuin-A precursor protein comprises 367 amino acids, which includes an 18-amino acid signal sequence (SS), a 282-amino acid A-chain, a 40-amino acid connecting peptide (CP), and a 27-amino acid B-chain. Structurally, the A-chain is composed of three distinct domains: two cystatin-like domains (D1 and D2) and a variable non-cystatin domain (D3a and D3b). Functionally, the D1 domain provides binding sites for calcium, transforming growth factor-beta (TGF-β), and bone morphogenetic proteins (BMPs); the D2 domain functions as a cysteine protease inhibitor; and the D3 domain facilitates interactions with cellular receptors, notably the insulin receptor 20,21.
Initially identified for its role in maintaining mineral homeostasis, Fetuin-A systemically inhibits ectopic calcification by forming soluble complexes with calcium and phosphate, thereby preventing their pathological deposition in soft tissues and the vasculature. This anti-calcific property is particularly clinically relevant in the context of chronic kidney disease and vascular calcification 22. However, accumulating evidence demonstrates that Fetuin-A also plays a significant role in the pathophysiology of insulin resistance and related cardiometabolic diseases 17,19,23. Furthermore, it actively participates in systemic inflammation and broader metabolic regulation 24. A primary mechanism underlying these metabolic effects is its ability to bind and inhibit the insulin receptor tyrosine kinase, thereby disrupting downstream insulin signaling cascades, including the IRS-1/PI3K/Akt pathway 25. By attenuating glucose uptake in peripheral tissues such as skeletal muscle and adipose tissue, this interference ultimately drives systemic insulin resistance and hyperglycemia.
Beyond its effects on insulin signaling, Fetuin-A regulates adipogenesis and hepatic lipid metabolism. It has been shown to suppress the secretion of adiponectin—an insulin-sensitizing adipokine—while concurrently promoting lipolysis and the release of free fatty acids from adipose tissue. Additionally, Fetuin-A is closely associated with non-alcoholic fatty liver disease (NAFLD) via mechanisms that inhibit fatty acid oxidation and promote de novo lipogenesis, both of which exacerbate hepatic steatosis 26. Emerging data also suggests that Fetuin-A acts as an innate immune modulator. In the presence of saturated fatty acids, Fetuin-A functions as an endogenous co-ligand for Toll-like receptor 4 (TLR4), triggering the synthesis of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β. This cascade exacerbates insulin resistance and perpetuates the chronic, low-grade inflammation characteristic of obesity and metabolic syndrome (MetS) 27.
Elevated circulating levels of Fetuin-A have been consistently documented in individuals with obesity, NAFLD, T2DM, and atherosclerotic CVD, suggesting that the glycoprotein serves as both a biomarker and an active effector of metabolic dysfunction.
Genetic Variants of AHSG and Fetuin-A Expression
Fetuin-A, a glycoprotein encoded by the AHSG gene on chromosome 3q27, exerts diverse functions in maintaining metabolic homeostasis. It is now widely recognized that genetic variations within this locus, particularly single nucleotide polymorphisms (SNPs), serve as critical determinants of circulating Fetuin-A concentrations, thereby modulating individual susceptibility to metabolic disorders such as insulin resistance, metabolic syndrome (MetS), and T2DM 17.
Prominent AHSG Gene Variants
a. rs4917 (Thr230Met or T230M)
Genetic variations within the AHSG gene have garnered significant interest due to their potential to influence metabolic disease pathogenesis. The rs4917 variant is among the most extensively studied SNPs associated with Fetuin-A. This polymorphism results in a missense mutation where methionine (M) replaces threonine (T) at amino acid position 230 (Thr230Met). Despite being a single amino acid substitution, this alteration can provoke profound physiological and biochemical repercussions 28.
At the molecular level, the T230M mutation may alter protein folding, stability, or post-translational modifications, subsequently impacting the secretion rate, half-life, or degradation dynamics of Fetuin-A. Even subtle perturbations in these properties can significantly modulate the protein's biological activity and systemic concentrations 29.
Functionally, this mutation has the potential to alter the interaction between Fetuin-A and key cellular receptors, notably Toll-like receptor 4 (TLR4) and the insulin receptor. Modifications in binding affinity or subsequent receptor activation could directly influence Fetuin-A's role in regulating lipid metabolism, triggering inflammatory cascades, and attenuating insulin signaling. Such mechanistic shifts may account for a substantial portion of the inter-individual variability observed in metabolic responses and disease pathogenesis 27.
Interestingly, functional genomics research reveals that individuals carrying the methionine (M) allele typically exhibit reduced circulating levels of Fetuin-A, which may confer a degree of metabolic protection. Lower Fetuin-A concentrations are correlated with enhanced insulin sensitivity, diminished systemic inflammation, and a reduced risk of hepatic steatosis—all of which mitigate the underlying risk factors associated with T2DM and cardiovascular disease (CVD) 26,30.
b. rs4918 (Thr256Ser)
The rs4918 single nucleotide polymorphism (SNP) is located in exon 7 of the AHSG gene. This variant constitutes a 767C>G missense mutation, resulting in the substitution of threonine for serine at amino acid position 256 (Thr256Ser). It is the most extensively studied AHSG polymorphism within the Indian population. This variant is significantly associated with T2DM and modulates circulating Fetuin-A levels. Specifically, the GG genotype of rs4918 has been significantly associated with reduced circulating Fetuin-A levels in patients with diabetic nephropathy 14. Conversely, the GG genotype has also demonstrated a protective effect, decreasing the risk of developing gestational diabetes mellitus (GDM) 31. Additionally, the G allele of the rs4918 SNP has shown a significant association with an increased risk of GDM (p=0.038). This study further reported that elevated Fetuin-A levels correlate with GDM and may contribute to underlying insulin resistance 32. Furthermore, current evidence suggests that the rs4918 variant does not exhibit a significant association with cardiovascular disease (CVD) risk 33.
c. rs2518136 (3′ UTR Variant)
Although it does not alter the amino acid sequence of the encoded protein, the rs2518136 SNP—located in the 3′ untranslated region (3′ UTR) of the AHSG gene—serves as a compelling example of how non-coding genetic variations can significantly impact gene expression and metabolic phenotypes. The rs2518136 variant of the AHSG gene has been significantly associated with an elevated risk of stroke 34 and T2DM 35.
d. rs2248690 (Upstream Promoter Variant)
The -799A/T (rs2248690) polymorphism, located in the promoter region of the AHSG gene, influences gene transcription and consequently alters circulating Fetuin-A levels 8,36. However, the genotype distribution of this specific variant has not shown any significant association with GDM 31.
Interestingly, investigations into the AHSG variants rs4917, rs2248690, and rs2518136 have not revealed any direct, significant associations with coronary atherosclerosis 37. Nevertheless, it has been reported that carriers of the T allele for the rs4917 variant exhibit elevated triglyceride levels and show a significant association with overweight and obesity 38. Given that obesity and dyslipidemia are primary risk factors for CVD, these genetic variations may indirectly influence long-term cardiovascular risk profiles 39.
Functional Consequences of AHSG Polymorphisms
These single nucleotide polymorphisms (SNPs) induce both quantitative and qualitative alterations in Fetuin-A expression, elevating their role from mere genetic markers to active functional modulators. Their pathophysiological influence manifests primarily in two distinct ways:
a. Plasma Level Variability
-
Research indicates that genetic variation within the
AHSG locus, particularly the rs4917 variant, accounts for up to 30–40% of the variance in circulating Fetuin-A levels. -
This fluctuation is significantly correlated with key clinical parameters, including liver fat content, fasting insulin levels, and the homeostasis model assessment for insulin resistance (HOMA-IR).
b. Metabolic Disease Risk
-
Candidate gene analyses and genome-wide association studies (GWAS) have robustly linked the rs4917 and rs2518136 variants to an elevated risk of T2DM, non-alcoholic fatty liver disease (NAFLD), atherosclerosis, and visceral obesity.
-
Individuals harboring these risk alleles frequently present with reduced high-density lipoprotein (HDL) cholesterol, elevated triglycerides, and heightened inflammatory markers, further substantiating the role of Fetuin-A as a critical nexus in cardiometabolic pathogenesis 26,40.
Ethnic and Population Variability
The allele frequencies and subsequent phenotypic impacts of AHSG variants exhibit significant heterogeneity across distinct global populations:
-
The rs4917 polymorphisms, which are associated with an elevated risk of T2DM, demonstrate a higher prevalence within East Asian populations.
-
In European cohorts,
AHSG polymorphisms have been independently associated with metabolic syndrome (MetS), irrespective of baseline obesity status. -
These epidemiological variations underscore the necessity of incorporating ethnicity-specific risk profiles when evaluating Fetuin-A as a diagnostic biomarker or a potential therapeutic target 9,14.
Clinical Relevance and Future Directions
Elucidating the functional impact of single nucleotide polymorphisms (SNPs) within the AHSG gene provides valuable clinical insights with substantial translational potential. For instance, the early identification of individuals at an elevated risk for insulin resistance, T2DM, and other metabolic disorders may be facilitated by genotyping key variants, such as the missense mutation rs4917 and the 3′ UTR variant rs2518136 7,38. These genetic markers hold promise as robust tools for risk stratification in both clinical and research settings. Looking forward, such genetic profiling could form the basis for personalized intervention strategies, enabling the customization of pharmacological therapies and lifestyle modifications according to an individual's AHSG genotype—thereby aligning with the broader paradigms of precision medicine 41. Furthermore, these insights pave the way for the development of targeted therapeutics designed to specifically modulate Fetuin-A function or AHSG expression. By targeting a fundamental biological driver of cardiometabolic disease, such interventions could offer a novel prophylactic or therapeutic approach to attenuate metabolic dysfunction, mitigate systemic inflammation, and preserve insulin sensitivity in genetically predisposed populations 17.
Mechanisms Linking Fetuin-A to T2DM
The pathogenesis of T2DM is characterized by an intricate interplay between genetic predispositions and metabolic derangements. A pivotal molecular mediator in this interaction is Fetuin-A, encoded by the AHSG gene. Fetuin-A contributes to the development of insulin resistance—a hallmark of T2DM—by perturbing three critical biological axes: the disruption of insulin signaling, the modulation of pro-inflammatory pathways, and the dysregulation of lipid metabolism.
Inhibition of Insulin Signalling
Insulin signaling is essential for maintaining systemic glucose homeostasis, as it regulates the uptake, storage, and utilization of glucose in key metabolic tissues, including the liver, skeletal muscle, and adipose tissue. Fetuin-A has emerged as a significant endogenous negative regulator of this pathway 42,43. By directly binding to the extracellular α-subunit of the insulin receptor (IR), Fetuin-A inhibits the receptor's ligand-induced autophosphorylation, a critical initial step required to trigger downstream intracellular signaling cascades. This interference subsequently suppresses the phosphatidylinositol 3-kinase (PI3K)/Akt signaling axis, thereby impairing the activation of insulin receptor substrates (IRS-1 and IRS-2). Consequently, the translocation of GLUT4-containing vesicles to the plasma membrane is markedly reduced, leading to diminished glucose uptake in insulin-sensitive tissues, predominantly skeletal muscle and adipose tissue 44.
This impairment in peripheral glucose disposal directly exacerbates postprandial hyperglycemia, a defining clinical feature of T2DM. Furthermore, Fetuin-A may also disrupt the mitogen-activated protein kinase (MAPK) pathway, which is integral to mitogenic signaling and cellular survival. The chronic inhibition of this pathway could compromise the viability and regenerative capacity of pancreatic β-cells, particularly under conditions of metabolic stress, thereby progressively worsening insulin deficiency over time 45,46,47.
Induction of Inflammation
Chronic, low-grade inflammation—often termed metaflammation—acts as both a cause and a consequence of insulin resistance. Fetuin-A exacerbates this bidirectional process by functioning as an endogenous ligand for Toll-like receptor 4 (TLR4). This interaction becomes particularly potent when Fetuin-A forms complexes with saturated fatty acids (SFAs), such as palmitic acid, which are typically elevated in states of obesity and T2DM (Figure 2). This binding event triggers several critical pro-inflammatory cascades:
-
Activation of the TLR4/MyD88/NF-κB signaling pathway: This prompts the nuclear translocation of NF-κB and the subsequent transcription of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1β.
-
Induction of macrophage infiltration: Fetuin-A drives the recruitment of macrophages into adipose tissue, thereby amplifying both local and systemic inflammatory responses.
-
Kinase-mediated IRS-1 phosphorylation: The activation of the JNK and IKKβ pathways leads to the phosphorylation of insulin receptor substrate-1 (IRS-1) at inhibitory serine residues, further attenuating downstream insulin signaling.
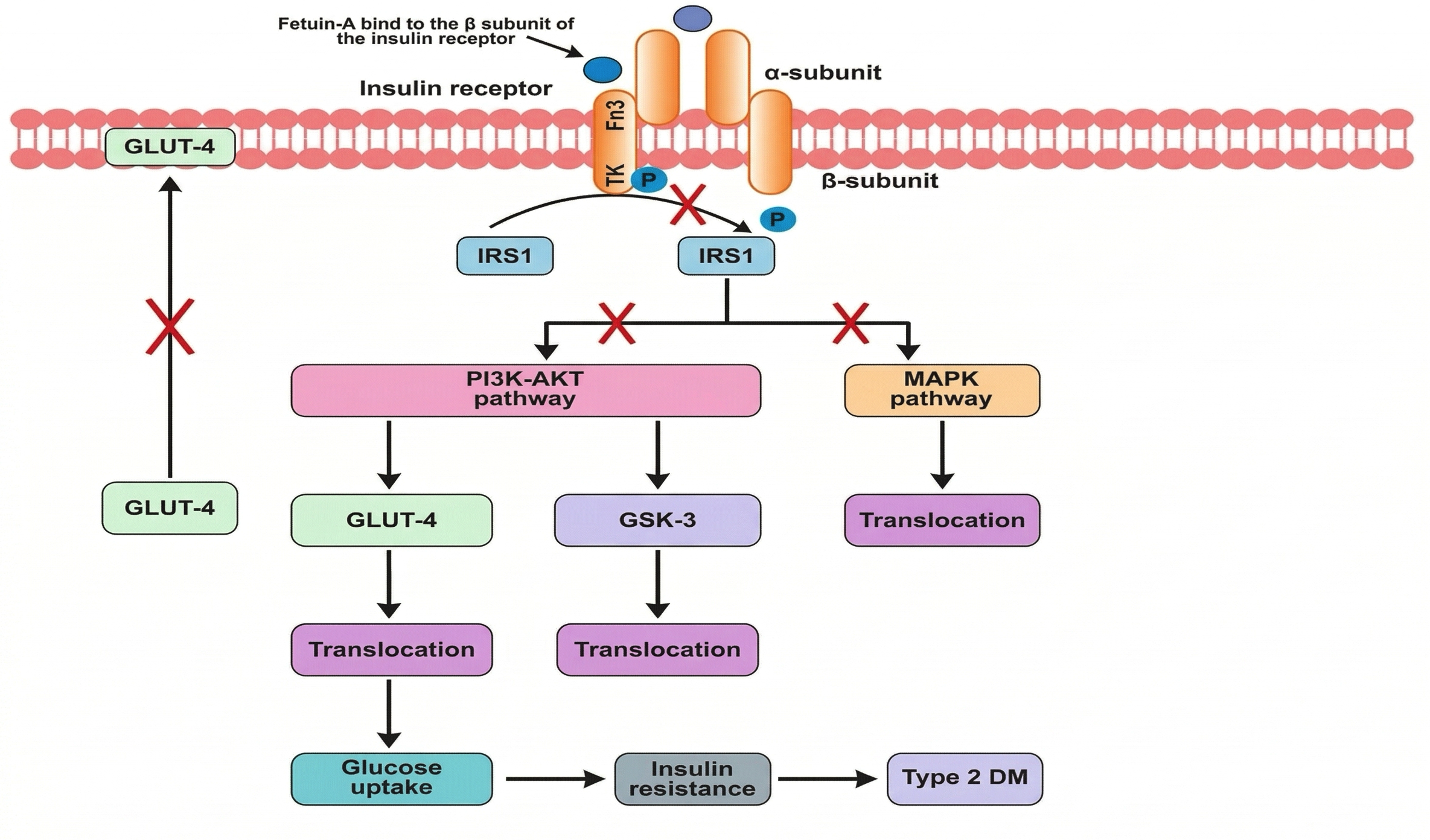
The role of fetuin-A in T2DM pathogenesis. Fetuin-A binds to the tandem Fn3 domain of the insulin receptor at a site distinct from the insulin-binding region. This interaction inhibits receptor autophosphorylation and disrupts downstream signaling by reducing tyrosine phosphorylation of IRS-1. Consequently, both the PI3K-Akt and MAPK pathways are attenuated, leading to decreased phosphorylation of Akt and GLUT4, impaired glucose uptake, and overall suppression of insulin signaling. These molecular events play a key role in the development of insulin resistance, a central mechanism in the pathogenesis of T2DM. DM, diabetes mellitus; Fn3, fibronectin type 3 domains; GLUT4, glucose transporter 4; GSK3, glycogen synthase kinase 3; IRS-1, insulin receptor substrate-1; MAPK, mitogen-activated protein kinase; T2DM, type 2 diabetes mellitus; TK, tyrosine kinase.
Even preceding the onset of overt hyperglycemia, Fetuin-A is frequently overexpressed in individuals with obesity, particularly those exhibiting visceral adiposity. In this context, it acts synergistically with circulating lipids to cultivate a pro-inflammatory milieu that drives the development of insulin resistance 45.
Dysregulation of Lipid Metabolism
Fetuin-A profoundly disrupts lipid metabolism through both central (hepatic) and peripheral (adipose and skeletal muscle) mechanisms:
a. Hepatic Lipid Accumulation and Steatosis
-
Fetuin-A promotes hepatic
de novo lipogenesis by upregulating lipogenic transcription factors, such as sterol regulatory element-binding protein 1c (SREBP-1c). -
Concurrently, it inhibits fatty acid oxidation by downregulating the expression of peroxisome proliferator-activated receptor alpha (PPAR-α) 48.
These combined actions precipitate intrahepatic lipid accumulation, fostering the progression of non-alcoholic fatty liver disease (NAFLD), which in turn exacerbates hepatic insulin resistance.
b. Adipocyte Dysfunction
Fetuin-A suppresses the expression and secretion of adiponectin, a potent anti-inflammatory adipokine responsible for enhancing insulin sensitivity and promoting fatty acid oxidation. Furthermore, Fetuin-A stimulates basal lipolysis within adipose tissue. This lipolytic activity results in elevated circulating levels of non-esterified fatty acids (NEFAs), which exert pronounced lipotoxic effects on vital metabolic organs, including the liver and the pancreas 49.
c. Ectopic Lipid Deposition and Lipotoxicity
Driven by elevated circulating NEFAs and diminished lipid oxidation, lipotoxicity manifests as the pathological accumulation of lipids in non-adipose tissues, most notably skeletal muscle and pancreatic β-cells. This ectopic lipid deposition significantly influences the pathophysiology of metabolic disorders by disrupting normal cellular homeostasis. In skeletal muscle, excessive intracellular lipid accumulation directly interferes with insulin signaling cascades, thereby exacerbating peripheral insulin resistance 50. In pancreatic β-cells, lipotoxic stress induces severe oxidative stress and mitochondrial dysfunction, ultimately culminating in defective insulin secretion. These deleterious effects compromise both the structural integrity and the secretory function of β-cells, thereby accelerating the transition from a state of compensated insulin resistance to overt T2DM. Consequently, lipotoxicity acts as a critical molecular nexus linking dysregulated lipid metabolism to the progressive loss of glycemic control 51,52.
Clinical and Experimental Evidence Linking Fetuin-A to Type 2 Diabetes Mellitus
A growing body of evidence derived from prospective cohort studies, genetic association analyses, and experimental models corroborates the role of Fetuin-A as a central mediator in the pathogenesis of T2DM. These findings not only reinforce the clinical utility of Fetuin-A as a predictive biomarker but also underscore its potential as a targeted therapeutic avenue in the management of metabolic disease 53.
Human Epidemiological Evidence
Several large-scale, longitudinal cohort studies have consistently demonstrated that elevated circulating Fetuin-A levels are highly predictive of incident T2DM, independent of conventional metabolic and lifestyle risk factors.
The EPIC-Potsdam Study
The European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam cohort represents one of the most prominent investigations in this domain. Comprising over 25,000 participants, this prospective study revealed that individuals with plasma Fetuin-A concentrations in the highest quartile exhibited a statistically significant 70% increased risk of developing incident T2DM compared to those in the lowest quartile. This elevated risk persisted even after robustly controlling for potential confounders, including age, sex, body mass index (BMI), physical activity, dietary habits, and smoking status. Crucially, the magnitude of this association remained unaffected by adjustments for circulating liver enzyme levels, strongly suggesting that Fetuin-A acts as an active pathogenic mediator of metabolic disease rather than merely serving as an epiphenomenal marker of hepatic dysfunction 15,54.
The Framingham Heart Study
The Framingham Offspring Study, a seminal longitudinal cohort, has provided robust genetic corroboration linking Fetuin-A to metabolic dysregulation 55. Polymorphisms within the AHSG gene—particularly the rs4917 and rs2248690 variants—were found to be strongly correlated with fasting insulin concentrations, HOMA-IR scores, and indices of adiposity. These genetic associations highlight a heritable component underpinning the metabolic consequences of Fetuin-A dysregulation, thereby supporting its conceptualization as a critical endophenotype that bridges genetic susceptibility with adverse clinical outcomes 17,19.
Interventional Studies: Impact of Lifestyle Modification
While observational research has consistently linked elevated Fetuin-A levels to metabolic dysfunction, interventional trials provide crucial causal evidence substantiating its active role in the pathogenesis of insulin resistance and T2DM. These studies highlight that lifestyle modifications not only improve conventional metabolic parameters but also directly modulate Fetuin-A expression and secretion 56.
It is well-established that dietary caloric restriction culminating in weight loss significantly reduces circulating Fetuin-A concentrations. Over a relatively short duration (typically 8 to 12 weeks), even moderate caloric deficits—independent of pharmacological interventions—can yield a 10–20% reduction in plasma Fetuin-A levels among obese or insulin-resistant individuals. This reduction frequently coincides with improved hepatic function, suggesting that alterations in liver metabolism are pivotal in regulating Fetuin-A synthesis 57,58. Similarly, exercise interventions, including both aerobic and resistance training, have been shown to significantly attenuate Fetuin-A levels independent of concurrent weight loss. This implies that physical activity modulates Fetuin-A through mechanisms distinct from mere adiposity reduction, such as the enhancement of hepatic lipid metabolism, the upregulation of anti-inflammatory cytokines, and the augmentation of insulin signaling pathways 56,59,60.
Crucially, these lifestyle-induced decrements in Fetuin-A levels consistently correlate with broad improvements in metabolic health. Participants typically exhibit reduced fasting glucose concentrations, enhanced insulin sensitivity, and attenuated systemic inflammation, as evidenced by diminished circulating levels of interleukin-6 (IL-6) and C-reactive protein (CRP) 61,62.
The reversibility of elevated Fetuin-A levels through targeted lifestyle interventions strongly supports its role as more than a mere passive biomarker. Rather, Fetuin-A operates as an active modulator of metabolic homeostasis, directly propagating insulin resistance and systemic inflammation 63,23. These findings underscore the therapeutic potential of modulating Fetuin-A via behavioral interventions, reaffirming its utility as both a therapeutic target and a prognostic monitoring tool in the prevention and management of cardiometabolic diseases.
Insights from Animal Models
Experimental animal studies provide crucial mechanistic validation of human epidemiological findings, facilitating a controlled examination of Fetuin-A’s biological functions.
a. Fetuin-A Knockout Models
Animal models have been instrumental in elucidating the molecular functions of Fetuin-A in metabolic regulation. Specifically, Fetuin-A knockout mice (Ahsg−/−), which are genetically engineered to lack the AHSG gene, have yielded critical insights into the hepatokine's role in obesity, insulin resistance, and chronic inflammation 27,64. Notably, Ahsg−/− mice exhibit markedly enhanced insulin sensitivity compared to their wild-type counterparts. This phenotype is characterized by improved glucose tolerance, reduced fasting insulin levels, and augmented insulin signaling within peripheral tissues, particularly skeletal muscle and adipose tissue. These metabolic benefits persist under both regular chow and high-fat diet (HFD) conditions, indicating that Fetuin-A deficiency confers robust metabolic protection independent of dietary stressors 27,64.
In addition to enhanced glucose metabolism, these mutant mice demonstrate remarkable resistance to diet-induced obesity. Despite equivalent caloric intake, they accumulate substantially less adipose tissue following prolonged high-fat feeding compared to wild-type controls. This observation suggests that Fetuin-A actively promotes adipogenesis and lipid storage, likely through the modulation of metabolic and inflammatory signaling pathways 65,66.
Crucially, Fetuin-A deficiency in these models is associated with attenuated Toll-like receptor 4 (TLR4) activation and a concomitant decrease in the production of downstream pro-inflammatory cytokines, including TNF-α and IL-6. These findings substantiate the hypothesis that Fetuin-A functions as an endogenous ligand for TLR4, thereby propagating the inflammatory cascades that precipitate insulin resistance. Consequently, these results reinforce the concept that Fetuin-A is not only a metabolic regulator but also a critical pro-inflammatory mediator linking liver-derived signals to systemic metabolic dysregulation 24,27.
Collectively, research utilizing Ahsg−/− mice provides compelling in vivo validation of the pathophysiological mechanisms proposed by human clinical and epidemiological investigations. Furthermore, these models establish a strong preclinical rationale for targeting Fetuin-A in the prevention and management of metabolic disorders 67.
b. Hepatic Overexpression Models
Complementing loss-of-function studies, transgenic mouse models engineered to overexpress Fetuin-A in the liver provide robust experimental evidence of its causal role in metabolic disease. By closely recapitulating the metabolic aberrations observed in human insulin resistance and T2DM, these gain-of-function models underscore the translational relevance of Fetuin-A as a primary pathogenic driver 68.
Hepatic Fetuin-A overexpression induces severe insulin resistance, predominantly affecting the liver and skeletal muscle—two primary sites of insulin action. This impairment in insulin signaling is characterized by diminished insulin-stimulated glucose uptake and attenuated Akt phosphorylation, indicative of disrupted insulin receptor activation 45,69. Metabolic profiling of these transgenic mice reveals elevated circulating levels of triglycerides and non-esterified fatty acids (NEFAs), mirroring the dyslipidemic phenotype frequently observed in patients with metabolic syndrome. These lipid abnormalities are likely mediated by Fetuin-A's disruptive effects on hepatic lipid metabolism and adipose tissue lipolysis 70.
Furthermore, hepatic overexpression of Fetuin-A induces a systemic pro-inflammatory state, evidenced by the upregulated expression of cytokines such as TNF-α and monocyte chemoattractant protein-1 (MCP-1), particularly within adipose tissue. These cytokines are established contributors to the chronic, low-grade inflammation that exacerbates insulin resistance and accelerates metabolic decline 71. Collectively, findings from transgenic overexpression models unequivocally demonstrate that Fetuin-A is an active mediator of metabolic dysfunction rather than a mere correlative marker. Its capacity to simultaneously impair insulin signaling, promote lipid dysregulation, and drive tissue inflammation highlights its central role in the pathogenesis of insulin resistance and T2DM 72.
Conclusion
Fetuin-A serves as a pivotal molecular mediator in the pathophysiology of T2DM, positioned at the nexus of genetic susceptibility and metabolic dysfunction. Variations within the AHSG gene—particularly single nucleotide polymorphisms (SNPs) such as rs4917 and rs2518136—significantly modulate the expression and functional activity of Fetuin-A, ultimately influencing an individual's susceptibility to insulin resistance and metabolic syndrome. Mechanistically, Fetuin-A drives insulin resistance through two primary avenues: the direct inhibition of insulin receptor autophosphorylation, which subsequently abrogates downstream insulin signaling cascades, and the activation of Toll-like receptor 4 (TLR4), which exacerbates systemic, low-grade inflammation. Furthermore, it profoundly alters lipid metabolism, promoting detrimental processes such as augmented lipolysis, ectopic lipid deposition, and hepatic steatosis.
Notably, this review highlights that the AHSG Thr256Ser (rs4918) variant is significantly associated with T2DM in South Indian populations. Interestingly, the homozygous mutant GG genotype of rs4918 appears to be responsible for reducing circulating Fetuin-A levels in this specific demographic. Despite this population-specific genetic nuance, elevated systemic Fetuin-A concentrations remain robustly associated with insulin resistance and the incidence of T2DM globally, underscoring its potential utility as a predictive biomarker for diabetes pathogenesis.
Abbreviations
3′ UTR: 3′ untranslated region; AHSG: Alpha-2-Heremans-Schmid glycoprotein; BMI: Body mass index; BMPs: Bone morphogenetic proteins; CP: Connecting peptide; CRP: C-reactive protein; CVD: Cardiovascular disease; DKD: Diabetic kidney disease; DM: Diabetes mellitus; EPIC: European Prospective Investigation into Cancer and Nutrition; Fn3: Fibronectin type 3 domains; GDM: Gestational diabetes mellitus; GLUT4: Glucose transporter 4; GSK3: Glycogen synthase kinase 3; GWAS: Genome-wide association studies; HDL: High-density lipoprotein; HFD: High-fat diet; HOMA-IR: Homeostasis model assessment for insulin resistance; IL-1β: Interleukin-1 beta; IL-6: Interleukin-6; IR: Insulin receptor; IRS-1: Insulin receptor substrate-1; IRS-2: Insulin receptor substrate-2; MAPK: Mitogen-activated protein kinase; MCP-1: Monocyte chemoattractant protein-1; MetS: Metabolic syndrome; NAFLD: Non-alcoholic fatty liver disease; NEFAs: Non-esterified fatty acids; PI3K: Phosphatidylinositol 3-kinase; PPAR-α: Peroxisome proliferator-activated receptor alpha; PRISMA: Preferred Reporting Items for Systematic Reviews and Meta-Analyses; SFAs: Saturated fatty acids; SNP: Single nucleotide polymorphism; SREBP-1c: Sterol regulatory element-binding protein 1c; SS: Signal sequence; T2DM: Type 2 Diabetes Mellitus; TGF-β: Transforming growth factor-beta; TK: Tyrosine kinase; TLR4: Toll-like receptor 4.
Acknowledgments
We are grateful to Prof. (Dr.) Abha Chandra, Dean, Integral Institute of Medical Sciences & Research (IIMSR), Integral University, Lucknow, India-226026 for the invaluable help and suggestions to write this article without any hindrance. I would like to acknowledge Integral University, Lucknow, India (IU/R&D/2025-MCN0001985).
Author’s contributions
All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have not used generative AI (a type of artificial intelligence technology that can produce various types of content including text, imagery, audio and synthetic data).
Competing interests
The authors declare that they have no competing interests.