
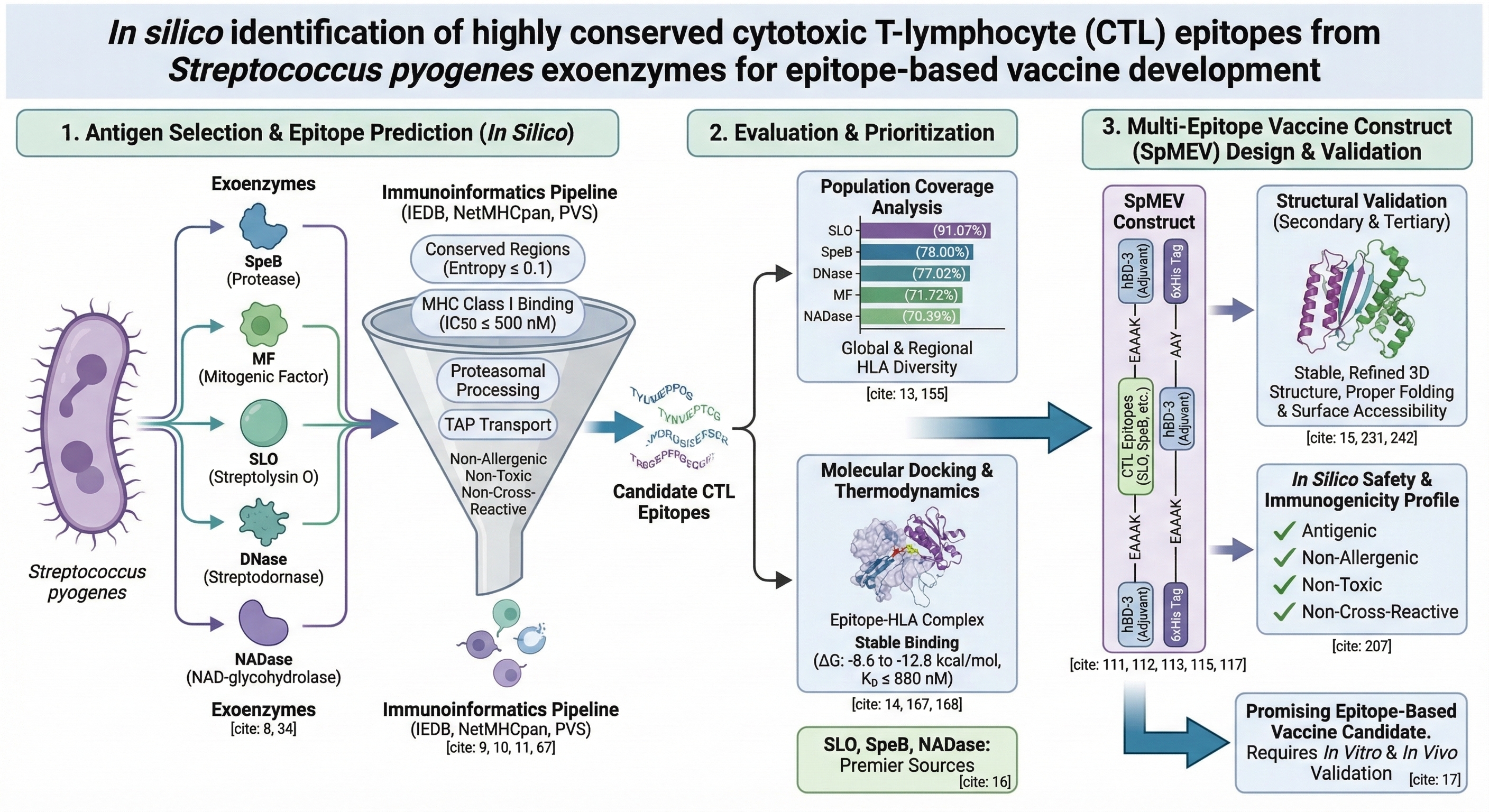

In silico identification of highly conserved cytotoxic T-lymphocyte (CTL) epitopes from Streptococcus pyogenes exoenzymes for epitope-based vaccine development
- Department of Biology, School of Science and Engineering, Ateneo de Manila University, Katipunan Ave., Quezon City, Philippines
- Department of Pharmaceutical Chemistry, College of Pharmacy, University of the Philippines Manila, Philippines
Abstract
Background: Streptococcus pyogenes represents a major global health concern attributable to its extensive repertoire of virulence factors, which contribute to severe diseases, most notably streptococcal pharyngitis. To facilitate the development of a safer, more targeted vaccine, this study employed a comprehensive in silico immunoinformatics approach to identify highly conserved cytotoxic T-lymphocyte (CTL) epitopes from five critical exoenzymes: SpeB, mitogenic factor (MF), streptolysin O (SLO), streptodornase (DNase), and NAD-glycohydrolase (NADase).
Methods: Conserved sequences were screened using the Protein Variability Server. CTL epitopes were predicted using Immune Epitope Database (IEDB) tools based on MHC class I binding affinity (IC50 500 nM), proteasomal processing, and TAP transport efficiency. Prioritization was given to non-allergenic, non-toxic, and non-cross-reactive epitopes to ensure safety. The selected epitopes were assembled into a multi-epitope vaccine construct, followed by the prediction, refinement, and validation of its secondary and tertiary structures.
Results: Population coverage analysis revealed high global HLA allele representation, with SLO-derived epitopes achieving the broadest worldwide population coverage (91.07%). Molecular docking and thermodynamic analysis validated strong, stable HLA-epitope interactions, with Delta G values ranging from -8.6 to -12. kcal/mol and dissociation constants (KD) ≤ 8.8×10-7M. Secondary structure prediction revealed a stable arrangement with balanced alpha-helices and beta-strands, while tertiary structure modeling and refinement confirmed proper folding and surface accessibility of the epitopes.
Conclusion: These findings highlight SLO, SpeB, and NADase as premier antigenic sources for epitope-based vaccine design. The resulting epitope set demonstrates promising immunogenic potential, safety, and population coverage, necessitating further in vitro and in vivo validation for streptococcal vaccine development.
Introduction
Streptococcus pyogenes (Group A Streptococcus, GAS) is a Gram-positive, beta-hemolytic bacterium.1 It is responsible for a wide range of diseases, from mild infections such as pharyngitis, cellulitis, and impetigo to severe invasive diseases including necrotizing fasciitis and streptococcal toxic shock syndrome.2 It also causes immune-mediated sequelae such as acute rheumatic fever (ARF) and rheumatic heart disease (RHD), which remain leading causes of cardiovascular morbidity in children and young adults in low- and middle-income countries. Globally, S. pyogenes accounts for approximately 616 million pharyngitis cases and 663,000 invasive infections annually, resulting in over 160,000 deaths.3 Despite this burden, no licensed vaccine currently exists against S. pyogenes. The high global incidence of S. pyogenes infections, increasing antimicrobial resistance to macrolides and clindamycin, and persistent challenges in disease surveillance, particularly in Southeast Asia, underscore the urgent need for alternative preventive strategies.4 Traditional vaccine development has been hindered by the pathogen’s antigenic diversity and the risk of autoimmune cross-reactivity with host tissues.
A promising approach against S. pyogenes is the development of epitope-based vaccines, which are highly targeted immunological constructs that employ short peptide sequences (epitopes) from key pathogen antigens to elicit specific immune responses.5 Unlike conventional vaccines that use whole organisms, inactivated pathogens, or full-length proteins, epitope-based vaccines focus on immunodominant regions of antigens to achieve protection.6 This approach offers several distinct advantages. In terms of safety, these vaccines exclude allergenic or toxic components of the pathogen, minimizing the risk of adverse effects.7 Specificity is enhanced by directing immune responses only toward conserved epitopes, reducing off-target effects and increasing the likelihood of effective protection across diverse strains of S. pyogenes. The design flexibility of these vaccines also allows for multivalent formulations containing multiple epitopes from different proteins, improving coverage against antigenic variability and serotype diversity.8 Finally, epitope selection can be tailored to suit different HLA haplotypes, improving global applicability.6
To support the development of a safe, specific, and broadly protective vaccine against S. pyogenes, this paper adopts an in silico immunoinformatics approach focused on an epitope-based vaccine design. This strategy targets five evolutionarily conserved and functionally significant exoenzymes namely, Streptococcal pyrogenic exotoxin B (SpeB), Mitogenic Factor (MF), Streptolysin O (SLO), NAD-glycohydrolase (NADase), and Streptodornase (DNase), selected based on their established roles in host-pathogen interactions and immune modulation. These exoenzymes are secreted virulence factors that contribute to the bacterium’s ability to invade host tissues, evade immune responses, and cause extensive cellular damage, making them strategic targets for cytotoxic T lymphocyte epitope identification.2 SpeB acts as a broad-spectrum protease capable of degrading host immunoglobulins, complement components, and extracellular matrix proteins, thereby compromising both innate and adaptive immune responses.9 MF functions as a superantigen, inducing non-specific T-cell activation and excessive pro-inflammatory cytokine release, which contributes to immune dysregulation and systemic inflammation.10 SLO is a pore-forming cytotoxin that disrupts host cellular membranes, resulting in immune cell lysis and tissue damage.11 NADase depletes intracellular NAD⁺, leading to energy loss and apoptotic death in immune cells, while DNase degrades neutrophil extracellular traps (NETs), facilitating immune evasion and enhanced bacterial dissemination.12
Given their conserved expression, extracellular localization, and immunological relevance, these exoenzymes represent rational targets for the identification of cytotoxic T lymphocyte (CTL) epitopes. This research aims to explore the potential of an immunoinformatics-guided approach in identifying conserved and immunogenic CTL epitopes from key S. pyogenes exoenzymes for epitope-based vaccine development. It focuses on selecting epitope candidates that demonstrate both broad population coverage and high safety profiles, including non-allergenic, non-toxic, and non-cross-reactive with the human proteins. In doing so, the study emphasizes the translational relevance of epitope-based vaccine strategies that address global HLA diversity and pathogen variability. Furthermore, it assesses the structural and thermodynamic plausibility of epitope-HLA interactions to support their suitability for downstream experimental validation. This work demonstrated how in silico approaches can be leveraged to inform the design of safe, effective, and globally relevant immunization strategies against S. pyogenes.
Materials and Methods
Protein Retrieval and Selection of Virulence Factors
Virulence-associated proteins were selected from the Virulence Factor Database (VFDB v.2025; ; accessed December 2025). The selected virulence factors included Streptococcal pyrogenic exotoxin B (SpeB), Mitogenic Factor (MF), Streptolysin O (SLO), Streptodornase (DNase), NAD-glycohydrolase (NADase), chosen on the basis of their established functional relevance in S. pyogenes pathogenicity. The corresponding amino acid sequences were retrieved in FASTA format from the NCBI Protein database (; accessed January 2026). A total of 100 non-redundant amino acid sequences were retrieved for each of the five major S. pyogenes exoenzymes from the NCBI Reference Sequence (RefSeq) database (Release 229, January 2026). This sample size was chosen to balance computational feasibility with sufficient representation of sequence diversity for conservation analysis. All available full-length, experimentally verified sequences were initially collected, excluding partial, hypothetical, or ambiguous entries. From this curated pool, redundancy was minimized using CD-HIT v.4.8.1 (; accessed January 2026) with a 90% identity cutoff, and 100 representative sequences per protein were selected to ensure a diverse yet manageable dataset. These sequences were subsequently used for multiple sequence alignment and epitope prediction analyses. The complete list of NCBI accession numbers for each protein is provided in the supplementary material.
Multiple Sequence Alignment and Conservation Analysis
Protein sequences corresponding to each Streptococcus pyogenes virulence factor were aligned using Clustal Omega v.1.2.4 (; accessed January 2026) to generate multiple sequence alignments (MSA). Sequence conservation was analyzed using the Protein Variability Server (PVS) (; accessed January 2026), where positional variability was quantified by the Shannon entropy (S) index. A fixed cutoff of S ≤ 0.1 was applied to define highly conserved residues, consistent with established thresholds in previous studies.13,14 Variable positions exceeding this threshold were masked and excluded, ensuring that only strongly conserved fragments (≥9 amino acids in length) were advanced for downstream MHC binding and immunogenicity analyses. The corresponding Shannon entropy profiles for each protein are provided in the supplementary material.
Epitope Prediction, MHC Binding Evaluation, and PTM Screening
Conserved regions from selected S. pyogenes virulence proteins were submitted to the Immune Epitope Database (IEDB) v2.28 (; accessed January 2026) for cytotoxic T-lymphocyte (CTL) epitope prediction. Predictions were performed using the NetMHCcons algorithm with default parameters, which integrates NetMHCpan v3.0 to assess MHC class I binding. Predicted epitopes were ranked based on combined scores incorporating MHC binding affinity, proteasomal cleavage likelihood, and TAP transport efficiency. A total of 36 globally and regionally common HLA class I alleles (HLA-A and HLA-B) were included to ensure broad population coverage. Predicted epitopes were filtered according to the following criteria: (1) MHC binding affinity IC₅₀ ≤ 500 nM, (2) proteasome cleavage score ≥ 1.0, (3) TAP transport score ≥ 1.0, and (4) non-redundant sequences only. For reference, NetMHCpan output scores typically range from 0 to 3.0, with higher values indicating stronger predicted immunogenic potential. Candidate peptides exceeding these thresholds were further screened for antigenicity, allergenicity, toxicity, and cross-reactivity using the IEDB analysis pipeline.
Because epitope-binding predictions are generated by machine learning models rather than conventional hypothesis-driven statistical tests, standard multiple-comparison corrections (e.g., FDR) are not directly applicable. To reduce false positives, only epitopes consistently identified as strong binders (IC₅₀ ≤ 500 nM) by two independent algorithms (NetMHCpan and NetCTL) were retained for downstream analysis. This concordance-based approach is commonly used in immunoinformatics studies and is supported by empirical evidence showing that epitopes predicted by multiple independent tools have a higher likelihood of representing true MHC binders.15
To further ensure structural stability, all candidate epitopes were assessed for post-translational modifications (PTMs) using UniProt v.2025_02 (). Potential PTM sites within the selected S. pyogenes virulence proteins were examined to ensure that predicted epitopes did not overlap with modified residues that could interfere with antigen processing or MHC binding. PTM annotations were retrieved from the UniProtKB database (release 2025_01; accessed January 2026) using the “Modified residue” and “Glycosylation site” features. Cross-verification using dbPTM version 2024 () confirmed the absence of annotated or predicted PTMs within the final epitope regions, ensuring structural and functional integrity of the selected peptides. Any epitope overlapping with modified residues was excluded to avoid processing interference.
Evaluation of Allergenicity, Toxicity, Cross-Reactivity, and Population Coverage
All predicted CTL epitopes were screened for allergenicity and toxicity. AllerTOP v.2.1 () and AllergenFP v.1.1 () apply auto-cross covariance (ACC) transformation and E-descriptor–based encoding of amino acids (hydrophobicity, size, and secondary-structure propensities) followed by machine-learning classification—k-nearest neighbor (AllerTOP) or Tanimoto-similarity (AllergenFP)—using a default probability cutoff of 0.5. Toxicity was predicted using ToxinPred2 (), an SVM-based model operating at its default confidence threshold. These tools exhibit an average false-positive rate of 10–15 %. Only epitopes predicted as non-allergenic and non-toxic by all tools were retained for downstream analyses.
To ensure non-reactivity with human proteins, each epitope was submitted to BLASTp analysis against the Homo sapiens (TaxID: 9606) proteome from the NCBI RefSeq protein database (Release 229, March 2025) using the NCBI BLAST tool (; accessed May 2025). Epitopes with E-values less than 1 were considered potentially cross-reactive and were excluded. Only those with E-values greater than 1 were retained as safe and non-homologous to human proteins.
The shortlisted epitopes were evaluated using the IEDB Population Coverage Tool (; accessed January 2026) to estimate their representation across HLA alleles in the Philippines, Southeast Asia, and global populations. This analysis provides point estimates of predicted population coverage, reflecting the computationally inferred potential immunogenicity of the selected epitopes across diverse genetic backgrounds. Confidence intervals were not calculated, as the current version of the IEDB tool does not support bootstrapping or uncertainty modeling. Consequently, the reported percentages should be interpreted as approximate estimates, which may vary with updates to allele frequency data or inclusion of additional population datasets.
Molecular Docking Analysis of Epitope-MHC Complexes
The top five epitopes exhibiting the highest predicted global population coverage were selected for molecular docking with their respective MHC class I alleles. Docking was performed using GalaxyPepDock (build 2023-05-29; ; accessed January 2026), a template-based peptide–protein docking tool. The three-dimensional structures of the corresponding HLA molecules were obtained from the Protein Data Bank (; accessed January 2026) and preprocessed in UCSF ChimeraX v.1.10, retaining only Chain A, which constitutes the peptide-binding domain. Non-relevant chains, ligands, and water molecules were removed prior to docking. All GalaxyPepDock runs used the server’s default parameters, which include automatic template selection from a database of experimentally determined peptide–protein complexes, rigid-body docking guided by interaction similarity, and flexible energy-based refinement of peptide and side-chain conformations. The server generates up to 10 candidate complex models per docking run, which are ranked by energy and interaction scores. Re-docking validation of native peptide–protein complexes was performed to confirm the reliability of the docking protocol, and all top-ranked poses were analyzed for structural consistency.
GalaxyPepDock employs a template-based deterministic docking algorithm, matching input sequences to structurally similar complexes rather than relying on stochastic sampling; therefore, random seeding is not applicable. All docking runs were conducted under uniform default parameters to ensure reproducibility and comparability of results. The resulting complexes were visually validated to confirm epitope localization within the peptide-binding groove, flanked by the characteristic α-helical regions. For each peptide–HLA pair, the top 10 docking models were retained to compute mean ± SD hydrogen bond statistics and evaluate interfacial interaction stability.
To evaluate the reliability of the docking protocol, a re-docking validation was conducted using the crystallized peptide–HLA-A*02:01 complex (PDB ID: 1I4F, 1.4 Å resolution). The native peptide was removed from the structure, and the receptor was used as the docking target. Docking was performed in GalaxyPepDock under identical parameters as in the main analysis. The resulting docked pose was superimposed onto the crystal structure using receptor Cα atoms, and the root-mean-square deviation (RMSD) between the docked and native peptide backbones was computed using PyMOL and UCSF ChimeraX. The re-docking yielded a Cα RMSD of 1.78 Å (all-atom RMSD = 2.31 Å), indicating excellent reproduction of the experimental binding conformation and confirming the reliability of the GalaxyPepDock protocol.
To evaluate the thermodynamic stability of the docked complexes, binding affinity was estimated using the PRODIGY server version 2.3.0 (; accessed January 2026). The Gibbs free energy of binding (ΔG) and the dissociation constant (K) at 37 °C were computed to quantify interaction strength. Complexes exhibiting lower ΔG values and K < 5 × 10⁻⁷ M were considered to possess strong and stable binding, indicative of favorable predicted immunogenic potential and suitability for vaccine development. The PRODIGY algorithm estimates ΔG based on interfacial contacts and solvent-accessible surface area, with an associated root-mean-square error (RMSE) of approximately ± 1.9 kcal/mol.
Design of the CTL-Based Multi-Epitope Vaccine Construct
A multi-epitope vaccine candidate (SpMEV) was designed by assembling computationally identified cytotoxic T lymphocyte (CTL) epitopes originating from different virulence-associated exoenzyme proteins of S. pyogenes. To preserve the immunogenicity of individual epitopes and minimize junctional immunogenicity, the CTL epitopes were concatenated using AAY linkers, which also facilitate effective antigen processing. To boost the overall immunostimulatory potential of the construct, Human β-defensin 3 (hBD-3) was incorporated as an adjuvant. hBD-3 is a host defense peptide that functions as a Toll-like receptor (TLR) agonist and possesses inherent antimicrobial as well as immune-enhancing properties. The full-length amino acid sequence of hBD-3 was fused at both N-and C-terminal end of the construct via a rigid EAAAK linker, ensuring structural stability and functional independence between the adjuvant and epitope array. A valine residue was added at the extreme N-terminus to enhance proteolytic resistance and prolong protein half-life within the host cell. For downstream purification applications, a C-terminal 6×His affinity tag was appended.
Safety Profiling: Allergenicity, Antigenicity, and Toxicity Assessment
The complete amino acid sequence of SpMEV was subjected to computational evaluation for antigenicity, allergenicity, and toxicity using established web-based prediction tools. Candidate suitability for further development was determined on the basis of being antigenic, non-allergenic, and non-toxic according to the default threshold scores provided by each algorithm.
Prediction of Physicochemical Characteristics
Key physicochemical descriptors of SpMEV including molecular weight, theoretical isoelectric point (pI), amino acid composition, estimated half-life, instability index, aliphatic index, and the grand average of hydropathicity (GRAVY) were calculated using the ProtParam tool on the ExPASy server v.3.0 (; accessed January 2026).
Structural Characterization: Secondary and Tertiary Structure Prediction
Secondary structure profiling of SpMEV was performed using PSIPRED v4.0 (; accessed June 2025), providing the predicted distribution of α-helices, β-strands, and random coils. PrDOS (; accessed January 2026) was employed to identify intrinsically disordered regions, while solvent accessibility of amino acid residues was analyzed through NetSurfP-3.0 (; accessed January 2026).
Three-dimensional structure modeling was conducted via a multi-template strategy. Initial structural models were generated using I-TASSER (; accessed January 2026) and GALAXY-TMB (; accessed January 2026), followed by refinement with GalaxyRefine2 (; accessed January 2026). Model validation was carried out through multiple approaches: (1) ERRAT (; accessed January 2026) to examine non-bonded atomic interactions); (2) ProSA-web (; accessed January 2026), for overall Z-score evaluation against crystallographic/NMR structures; and (3) PROCHECK (; accessed January 2026) to assess stereochemical quality via Ramachandran plot statistics.
The final validated 3D structure of SpMEV was visualized in UCSF ChimeraX v.1.10 () where the structural organization and surface accessibility of predicted CTL epitopes were examined to ensure adequate presentation for immune recognition.
Results
Five key S. pyogenes exoenzymes—SpeB, MF, DNase, SLO, and NADase—were selected as target antigens based on their central roles in virulence and immune evasion. These proteins facilitate host invasion and immune suppression through mechanisms such as proteolytic degradation (SpeB)12, superantigenic activation (MF)10, NET degradation (DNase)17, pore formation (SLO)18, and NAD⁺ depletion (NADase)19. Their conserved, surface-exposed, and immunogenic nature makes them strong candidates for cytotoxic T lymphocyte (CTL) epitope-based vaccine design.
Identification and Immunogenic Profiling of Conserved Cytotoxic Epitopes from S. pyogenes Exoenzymes
To ensure broad protection across diverse S. pyogenes strains, conserved antigenic regions were identified for each target protein using the Protein Variability Server, retaining fragments with Shannon entropy ≤ 0.1 and a minimum length of nine (9) amino acids. Low entropy indicates strong evolutionary conservation, and a nine-residue length corresponds to the optimal size for MHC class I binding. The ≤ 0.1 cutoff reflects minimal sequence variability and is widely used in immunoinformatics to define highly conserved regions suitable for vaccine targets. This approach prioritizes functionally constrained regions with high cross-strain immunogenic potential, reducing the risk of immune escape in bacterial pathogens.13,14
Predicted CTL epitopes from these conserved regions were screened for MHC class I affinity using NetMHCpan within IEDB. Peptides with IC₅₀ ≤ 500 nM were classified as strong binders, and only those with favorable proteasomal cleavage and TAP transport scores (≥ 1.0) were retained, ensuring their likelihood of natural processing and presentation.20,21 To maintain biological relevance, predicted epitopes were cross-checked with post-translational modification (PTM) sites from UniProt, as PTMs can alter peptide stability and HLA binding.22 Epitopes overlapping PTM sites were excluded, particularly for SpeB, MF, and SLO.
Safety assessment included filtering for allergenicity, toxicity, and human cross-reactivity, minimizing the risk of off-target immune responses. Allergenicity was assessed via AllerTOP v.2.1 and AllergenFP v.1.1, and peptides flagged as probable allergens by either tool were excluded to avoid IgE-mediated hypersensitivity responses, commonly elicited by small antigens capable of cross-linking IgE on mast cells.23 ToxinPred2 was used to assess peptide toxicity based on amino acid composition and motifs, retaining only non-toxic sequences to minimize off-target or cytotoxic effects.24 To prevent autoimmune risk, all non-toxic epitopes were screened against the human proteome using BLASTp, applying a stringent E-value cut-off (>1) to eliminate sequences with significant similarity to endogenous human proteins. Peptides exhibiting E-values less than 1 were flagged as potentially cross-reactive and excluded, as such homology may trigger autoreactive T-cell responses and increase the risk of autoimmune complications.25 The retained epitopes exhibited E-values ranging from 1.2 to 73, confirming the absence of relevant homology and minimizing the potential for cross-reactive autoimmunity.
Among the five exoenzymes, SLO yielded the highest number of conserved and immunogenic CTL epitopes (n = 35), underscoring its evolutionary stability and immunodominance across >98% of S. pyogenes isolates.26 SpeB (n = 19) and DNase (n = 16) contributed epitopes consistent with their established roles in immune evasion via proteolysis and NET degradation, respectively. NADase (n = 14) and MF (n = 10) provided complementary epitopes associated with intracellular survival and superantigenic activity. The final list of epitopes per virulence factor is summarized in Table 1.
Summary of cytotoxic T-lymphocytes epitopes containing at least two HLA binders classified as nonallergenic, nontoxic, and non-cross-reactive
| Virulence Factors | CTL Predicted Epitopes |
|---|---|
| SpeB | 19 |
| MF | 10 |
| DNAse | 16 |
| SLO | 35 |
| NADase | 14 |
Evaluation of HLA Binding Promiscuity and Population Coverage
In this study, the final CTL epitopes were analyzed using the IEDB Population Coverage Tool, which integrates experimentally verified HLA frequency data across diverse ethnic groups. Results revealed that SLO-derived epitopes achieved the highest global coverage (91.07%), followed by SpeB (78.00%), DNase (77.02%), MF (71.72%), and NADase (70.39%) (Table 2). These values represent the proportion of the global population predicted to mount a CTL response to at least one epitope from each antigenic source.
Population coverage of cytotoxic T-lymphocytes epitopes
| Virulence Factors | Philippines | Southeast Asia | World |
|---|---|---|---|
| SpeB | 62.79% | 79.18% | 78.00% |
| MF | 13.51% | 58.67% | 71.72% |
| DNAse | 13.51% | 36.81% | 77.02% |
| SLO | 69.86% | 83.05% | 91.07% |
| NADase | 69.86% | 75.53% | 70.39% |
Regional analysis further revealed allele-specific variations influencing local immunogenic potential. Using IEDB-reported HLA class I frequencies (11 documented alleles for the Philippines), DNase and MF epitopes showed limited combined coverage (13.51%), reflecting regional allele bias toward non-dominant supertypes.27 In contrast, SLO and NADase epitopes retained high compatibility (69.86%) with locally prevalent alleles, suggesting favorable regional immunogenicity.
Structural Binding Validation via Molecular Docking
Molecular docking was employed to evaluate the structural compatibility and binding strength of the top five CTL epitopes from S. pyogenes exoenzymes (SpeB, MF, SLO, DNase, and NADase) with their respective HLA class I alleles. These epitopes were pre-screened based on high MHC affinity (IC₅₀ ≤ 500 nM), non-allergenicity, non-toxicity, and HLA-binding promiscuity, ensuring immunologically robust candidates for downstream validation.
All epitopes were found to occupy the canonical MHC class I peptide-binding cleft, forming stabilizing hydrophobic and hydrogen bonds consistent with known HLA–peptide interactions (Figure 1). Anchor residues located at positions 2, 5, and the C-terminus mediated the strongest contacts, reinforcing peptide stability and proper groove alignment.28 No structural clashes or misorientations were observed, and all peptides adopted TCR-accessible conformations, supporting their potential for effective antigen presentation and CD8⁺ T-cell activation.29
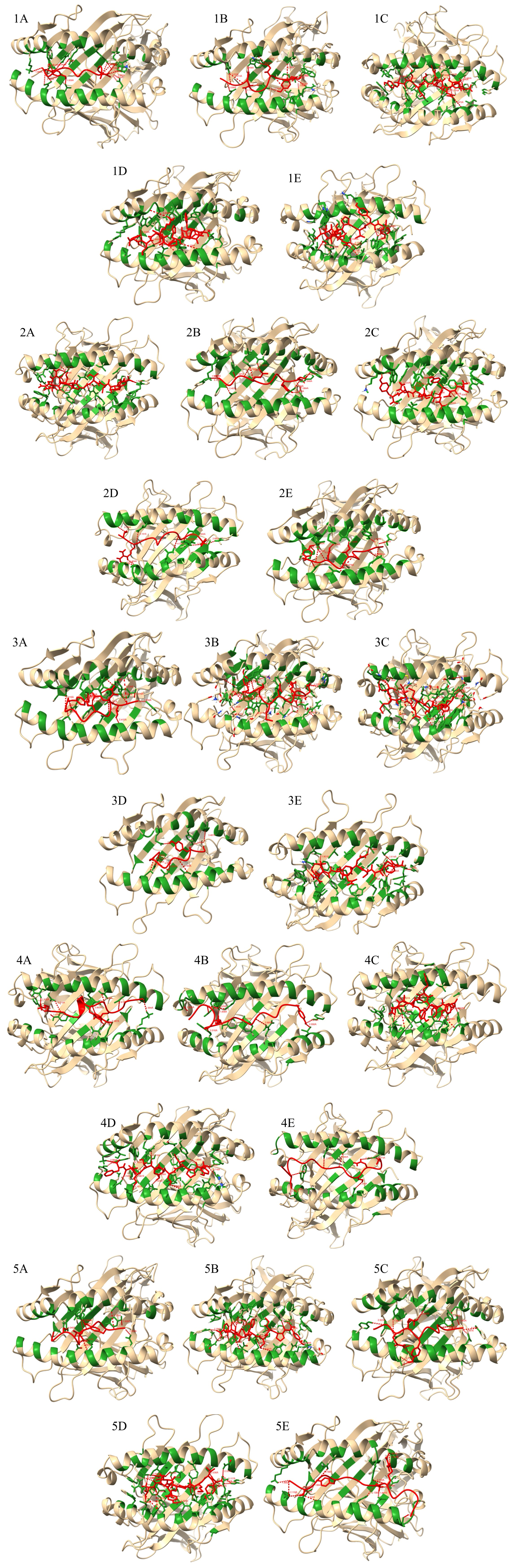
Representative molecular docking interactions between cytotoxic T lymphocyte (CTL) epitopes derived from exoenzyme proteins and their corresponding HLA molecules. The panels illustrate docked complexes of (A) SpeB, (B) MF, (C) DNase, (D) SLO, and (E) NADase epitopes within the peptide-binding groove of their respective HLA structures. The green-colored region represents the 5 Å interaction interface between each epitope and the HLA binding pocket, highlighting the spatial proximity that facilitates molecular recognition. Red dotted lines denote hydrogen bond interactions, with corresponding bond lengths indicated in the figure. These docking visualizations depict the key molecular interactions stabilizing epitope–HLA binding, providing structural insights into antigen presentation and potential immunogenicity.
To assess thermodynamic favorability, binding free energy (ΔG) and dissociation constants (K) were calculated using the PRODIGY server. All epitope–HLA complexes exhibited negative ΔG values (≤ –8.6 kcal/mol) and submicromolar K values (≤ 880 nM), indicating spontaneous and stable binding under physiological conditions (Table 3). The NADase–HLA-B15:01–TQYTESMVY complex displayed the strongest interaction (ΔG = –12.8 kcal/mol; K = 0.99 nM), while the DNase–HLA-B15:01–KDMIDMSAGY complex exhibited the weakest yet favorable binding (ΔG = –8.6 kcal/mol; K = 880 nM). These values fall within the range expected for stable protein–peptide complexes, validating the docking accuracy and biological relevance of the modeled interactions.
KD and ΔG values of the 10 best-ranked HLA–epitope complex models based on predicted binding affinity
| Virulence Factors | HLA-epitope complex | ΔG (kcal/mole) | KD (nM) |
|---|---|---|---|
| SpeB | A1: HLA-A*11:01 - ATATAQIMKY | -10.6 ± 0.2 | 32 ± 15 |
| A2: HLA-A*68:01- YTYTLSSNNPY | -9.9 ± 0.4 | 110 ± 38 | |
| A3: HLA-A*11:01 - YTLSSNNPY | -10.5 ± 0.1 | 38 ± 11 | |
| A4: HLA-A*24:02- YFNHPKNLF | -10.2 ± 0.1 | 69 ± 4 | |
| A5: HLA-A*29:0 - LSQNQPVYY | -9.8 ± 0.2 | 110 ± 22 | |
| MF | B1: HLA-A*03:01- RTARGTLTY | -9.8 ± 1.1 | 130 ± 54 |
| B2: HLA-A*03:01 - LTYANVEGSY | -9.1 ± 0.3 | 380 ± 61 | |
| B3: HLA-A*03:01 - LVYNTANGY | -9.3 ± 0.2 | 280 ± 17 | |
| B4: HLA-B*58:01 - TRTARGTLTY | -10.8 ± 0.5 | 25 ± 9 | |
| B5: HLA-A*03:01 - VLVYNTANGY | -9.7 ± 0.3 | 140 ± 16 | |
| DNase | C1: HLA-B*15:01 - ITENTSSTIY | -8.8 ± 0.2 | 660 ± 49 |
| C2: HLA-B*15:01 - KDMIDMSAGY | -8.6 ± 0.1 | 880 ± 73 | |
| C3: HLA-B*15:01 - DMIDMSAGY | -9.4 ± 0.2 | 220 ± 39 | |
| C4: HLA-B*15:01 - HVYYKATPVY | -11.6 ± 0.2 | 6.7 ± 1.2 | |
| C5: HLA-B*15:01 - RVFNNVAGF | -9.5 ± 0.6 | 200 ± 11 | |
| SLO | D1: HLA-A*03:01 - KVMIAAYKQIFY | -11.2 ± 2.4 | 13 ± 0.8 |
| D2: HLA-A*03:01 - ATFSRKNPAYPISY | -10.1 ± 1.8 | 75 ± 15 | |
| D3: HLA-A*03:01 - AAYKQIFY | -9.0 ± 1.9 | 480 ± 44 | |
| D4: HLA-B*07:02 - YPISYTSVF | -9.1 ± 2.2 | 370 ± 23 | |
| D5: HLA-B*58:01 - VSNEAPPLFVSNVAY | -10.4 ± 2.0 | 43 ± 18 | |
| NADase | E1: HLA-A*24:02 - AYPISYTSVF | -10.2 ± 0.3 | 61 ± 31 |
| E2: HLA-B*07:02 - YPISYTSVF | -9.4 ± 0.1 | 240 ± 25 | |
| E3: HLA-A*24:02 - VMIAAYKQIF | -9.6 ± 0.2 | 180 ± 13 | |
| E4: HLA-B*15:01 - TQYTESMVY | -12.8 ± 0.2 | 0.9 ± 0.1 | |
| E5: HLA-A*11:01 - ATFSRKNPAYPISY | -12.4 ± 0.2 | 1.8 ± 0.3 |
A strong inverse correlation was observed between the predicted Gibbs free energy of binding (ΔG) and the dissociation constant (K) across all epitope–HLA complexes (Figure 2). Complexes with more negative ΔG values exhibited lower K values, reflecting higher binding affinity and complex stability. Among the analyzed virulence factors, epitopes derived from NADase, SLO, and SpeB demonstrated the strongest binding interactions (ΔG ≤ –11 kcal/mol; K in the nanomolar range), supporting their immunogenic potential.
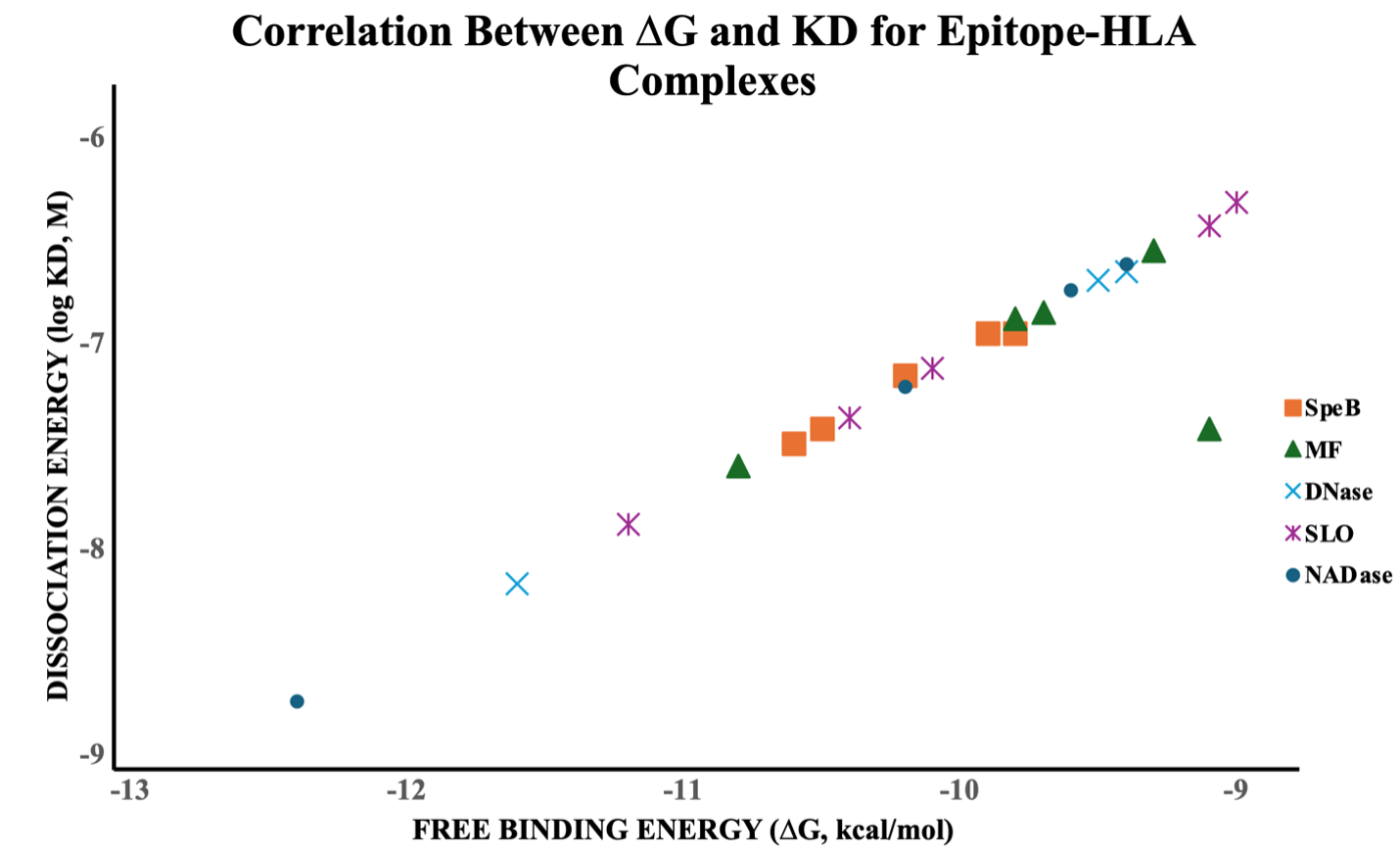
Correlation between Binding Free Energy (ΔG) and Dissociation Constant (KD) for Epitope–HLA Complexes. The scatter plot illustrates the relationship between the calculated free binding energy (ΔG, kcal/mol) and the corresponding dissociation constant (KD, M) of predicted epitope–HLA complexes derived from Streptococcus pyogenes virulence factors, including SpeB, MF, DNase, SLO, and NADase. Each point represents the mean binding energy and dissociation constant derived from the top docking pose of each epitope–HLA pair. Distinct colors and marker shapes denote different virulence proteins, allowing comparison of binding affinities across antigenic groups. The strong inverse correlation between ΔG and log KD indicates that lower binding energies correspond to tighter binding affinities, consistent with thermodynamic principles governing ligand–receptor interactions. These results confirm the reliability of the docking-derived affinity estimations and support the selection of epitopes with superior binding stability for further immunogenicity evaluation.
Hydrogen Bond Geometry and Stability of HLA–Epitope Complexes
The geometric parameters of hydrogen bonds provide critical insights into their strength and role in molecular recognition. Empirical studies have established that donor–acceptor (D···A) distances of 2.7–3.3 Å typically denote stable hydrogen bonds, while distances below 3.0 Å indicate stronger and more favorable interactions.30 Likewise, hydrogen–acceptor (H···A) distances of 1.6–2.5 Å are characteristic of stable hydrogen bonding, with shorter values in the 1.6–2.2 Å range representing near-optimal interactions when complemented by favorable bond angles. These geometric parameters are particularly significant in peptide–MHC (pMHC) systems, where robust hydrogen bonding helps anchor conserved peptide residues within the MHC binding groove, promoting specificity and long-term complex stability.30
As summarized in Table 4, all modeled complexes displayed extensive hydrogen bonding networks, ranging from 10 to 30 hydrogen bonds per complex. Donor–acceptor distances spanned approximately 2.70–4.92 Å, and hydrogen–acceptor distances ranged from 1.84–3.95 Å. The lower-bound values fall within the strong hydrogen bond regime, especially those near 1.84–2.0 Å (H···A) and 2.7–3.0 Å (D···A), suggesting favorable enthalpic contributions to binding affinity. Notably, DNase-derived epitopes in complexes C2 and C3 exhibited the highest number of hydrogen bonds (28 and 30, respectively), indicating highly stabilized interactions with their respective HLA alleles. Such dense hydrogen bond networks are known to increase complex half-life and enhance immunogenicity in pMHC systems, as they promote stable epitope anchoring and efficient presentation to cytotoxic T lymphocytes.27 Conversely, interactions near the upper distance limits (up to ~3.9 Å for H···A and ~4.9 Å for D···A) likely correspond to weaker or water-mediated hydrogen bonds. While individually less stabilizing, these interactions contribute to the overall orientation, flexibility, and conformational adaptability necessary for effective T-cell receptor recognition.
Hydrogen bond profiles and geometric parameters of epitope–HLA complexes
| Exoenzymes protein | HLA-epitope complex | No. of H-bonds | H---acceptor distance (Å) | D---acceptor distance (Å) |
|---|---|---|---|---|
| SpeB | A1: HLA-A*11:01 - ATATAQIMKY | 16 ± 1 | 1.916-3.947 | 2.704-4.917 |
| A2: HLA-A*68:01- YTYTLSSNNPY | 16 ± 1 | 1.948-4.220 | 2.833-4.980 | |
| A3: HLA-A*11:01 - YTLSSNNPY | 15 ± 2 | 1.866-4.502 | 2.781-5.295 | |
| A4: HLA-A*24:02- YFNHPKNLF | 11 ± 1 | 2.003-4.158 | 2.870-4.896 | |
| A5: HLA-A*29:0 - LSQNQPVYY | 12 ± 1 | 1.895-3.911 | 2.845-4.705 | |
| MF | B1: HLA-A*03:01- RTARGTLTY | 11 ± 2 | 1.909-3.650 | 2.849-4.529 |
| B2: HLA-A*03:01 - LTYANVEGSY | 13 ± 3 | 1.884-3.650 | 2.816-4.598 | |
| B3: HLA-A*03:01 - LVYNTANGY | 11 ± 1 | 1.851-4.126 | 2.749-4.921 | |
| B4: HLA-B*58:01 - TRTARGTLTY | 11 ± 1 | 1.899-4.336 | 2.777-5.020 | |
| B5: HLA-A*03:01 - VLVYNTANGY | 12 ± 2 | 1.950-4.678 | 2.745-4.936 | |
| DNase | C1: HLA-B*15:01 - ITENTSSTIY | 17 ± 3 | 1.937-3.536 | 2.864-5.134 |
| C2: HLA-B*15:01 - KDMIDMSAGY | 28 ± 3 | 1.791-3.725 | 2.703-5.378 | |
| C3: HLA-B*15:01 - DMIDMSAGY | 30 ± 4 | 1.902-4.202 | 2.726-5.003 | |
| C4: HLA-B*15:01 - HVYYKATPVY | 10 ± 4 | 1.906-4.523 | 2.833-4.970 | |
| C5: HLA-B*15:01 - RVFNNVAGF | 10 ± 4 | 1.901-3.535 | 2.908-4.414 | |
| SLO | D1: HLA-A*03:01 - KVMIAAYKQIFY | 11 ± 2 | 1.900-3.540 | 2.721-4.648 |
| D2: HLA-A*03:01 - ATFSRKNPAYPISY | 10 ± 2 | 1.858-4.325 | 2.830-5.034 | |
| D3: HLA-A*03:01 - AAYKQIFY | 11 ± 1 | 1.990-3.909 | 2.797-5.191 | |
| D4: HLA-B*07:02 - YPISYTSVF | 14 ± 5 | 1.915-3.716 | 2.786-4.566 | |
| D5: HLA-B*58:01 - VSNEAPPLFVSNVAY | 8 ± 5 | 1.948-3.690 | 2.883-4.389 | |
| NADase | E1: HLA-A*24:02 - AYPISYTSVF | 10 ± 3 | 1.926-4.040 | 2.913-4.766 |
| E2: HLA-B*07:02 - YPISYTSVF | 19 ± 3 | 1.847-4.316 | 2.806-5.138 | |
| E3: HLA-A*24:02 - VMIAAYKQIF | 14 ± 2 | 1.962-4.147 | 2.858-5.071 | |
| E4: HLA-B*15:01 - TQYTESMVY | 22 ± 1 | 1.943-4.317 | 2.846-5.587 | |
| E5: HLA-A*11:01 - ATFSRKNPAYPISY | 15 ± 1 | 1.939-3.738 | 2.755-4.545 |
SpMEV Construct Design and Structural Features
A multi-epitope peptide vaccine candidate against Streptococcus pyogenes (SpMEV) was developed by systematically integrating six cytotoxic T-lymphocyte (CTL) epitopes that exhibited the most favorable thermodynamic characteristics, specifically the lowest predicted Gibbs free energy (ΔG) and dissociation constant (K) values. These epitopes, derived from immunodominant S. pyogenes exoenzymes, were arranged in tandem and connected by AAY linkers.31 The AAY linker facilitates proteasomal cleavage and efficient presentation via the MHC class I pathway32, ensuring proper processing of each epitope while reducing the formation of junctional epitopes that could compromise immunogenicity. While this design minimizes the formation of junctional epitopes, it is important to note that novel junctional sequences may still arise and influence epitope processing or immunodominance, warranting future experimental validation. The construct was deliberately designed as a CTL-focused vaccine to stimulate robust cell-mediated immunity against S. pyogenes, particularly targeting the intracellular persistence phase of infection.33
To enhance immunostimulatory potential, human β-defensin-3 (hBD-3) was incorporated at both the N- and C-termini of the construct as an intrinsic adjuvant. hBD-3 was selected due to its documented antimicrobial activity and its capacity to promote antigen presentation and dendritic-cell maturation.34 Previous studies have reported hBD-3–mediated maturation of Langerhans-like dendritic cells and adjuvant effects in cutaneous vaccination models, whereas other in vivo findings indicate context-dependent immunosuppression, modulation of innate signaling pathways, and potential pro-inflammatory responses.35,36
Rigid EAAAK linkers were employed to join the hBD-3 adjuvant domains to the CTL epitope cluster, ensuring structural independence, optimal spatial separation, and reduced interference between the epitopes and adjuvant segments. A valine residue was appended at the C-terminus to improve structural stability and protease resistance, extending the potential half-life of the construct. In addition, a polyhistidine (6×His) tag was introduced to facilitate downstream purification via affinity chromatography, improving the construct’s feasibility for recombinant expression and large-scale production. The schematic diagram of the final SpMEV construct (Figure 3) consisted of 170 amino acids organized in a rational architecture balancing immunogenicity, stability, and manufacturability.
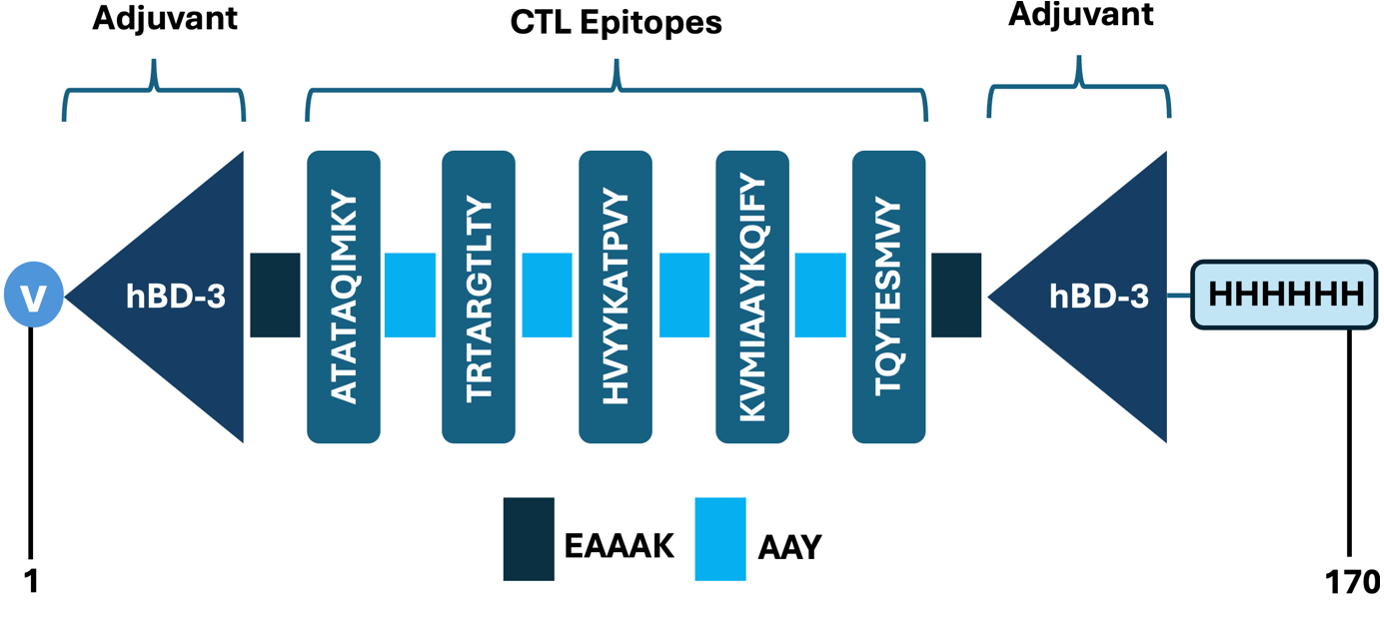
Schematic representation of the SpMEV construct
In Silico Characterization of Immunogenic and Safety Profiles of the SpMEV Construct
The designed Streptococcus pyogenes multi-epitope vaccine (SpMEV) construct was subjected to immunoinformatic evaluation to assess its antigenicity, allergenicity, toxicity, and cross-reactivity. Antigenicity analysis using VaxiJen yielded a score of 0.5387, which surpasses the default threshold of 0.4, while ANTIGENpro predicted a significantly higher value of 0.9137. These results suggest that the construct possesses strong intrinsic immunogenic potential, an essential feature for eliciting protective immune responses.23
Allergenicity prediction indicated that the construct is a probable non-allergen, supported by a Tanimoto coefficient of 0.4368 when compared to the most similar human protein, the cystic fibrosis transmembrane conductance regulator (CFTR). This relatively low similarity index supports its classification as non-allergenic, reducing the likelihood of hypersensitivity reactions. Importantly, AllerCatPro also classified the construct as a probable non-allergen, with no significant sequence similarity detected at the stringent E-value threshold of 0.001, further strengthening confidence in its safety profile.
Further evaluation using ToxinPred2 classified the construct as non-toxic, strengthening its safety profile for potential in vivo applications. Cross-reactivity screening also confirmed that the construct does not share significant similarity with human proteins, thereby minimizing the risk of autoimmune responses. Finally, these results indicate that the SpMEV construct is predicted to be highly antigenic, non-allergenic, non-toxic, and non-cross-reactive, supporting its suitability as a safe and effective vaccine candidate. Structural validation (secondary and tertiary) was subsequently conducted to ensure the stability and reliability of the construct.
Physicochemical properties of the SpMEV construct
The final SpMEV construct comprises 170 amino acids, with a theoretical molecular weight of 19.37 kDa and an isoelectric point (pI) of 9.89, suggesting a basic and predominantly cationic protein at physiological pH. The overall amino acid composition indicates enrichment in positively charged residues, contributing to a net charge favorable for electrostatic interactions with antigen-presenting cells. The predicted extinction coefficient (26,080 M⁻¹ cm⁻¹) and instability index (39.97) indicate good structural stability, while an aliphatic index of 65.59 and a GRAVY score of 0.47 suggest moderate thermostability and hydrophilicity, respectively—properties generally consistent with soluble and immunogenic recombinant proteins. The estimated half-lives across mammalian, yeast, and bacterial systems (≥ 10 h) further support the construct’s potential suitability for heterologous expression. The detailed ProtParam outputs (amino acid composition, charge distribution, extinction coefficient assumptions, and atomic composition) are provided in supplementary material. These summarized physicochemical characteristics collectively indicate that the SpMEV construct is predicted to be a stable, soluble, and moderately thermostable vaccine candidate suitable for downstream expression and validation.
Secondary Structure of the SpMEV Construct
The secondary structure profile of the designed SPMEV vaccine revealed a favorable balance of α-helices, β-strands, and coil regions (Figure 4). From an immunological perspective, such structural diversity is advantageous because it provides both rigidity and flexibility within the vaccine construct. The α-helical and β-strand elements contribute to the stability of the overall fold, ensuring that epitopes are maintained in a defined conformation that can be consistently recognized by the immune system. In contrast, coil regions provide structural adaptability, often facilitating the surface presentation of epitopes to major histocompatibility complex (MHC) molecules. This combination of ordered and flexible domains reflects a rational design principle in epitope-based vaccines, where stability and accessibility must be carefully balanced.37
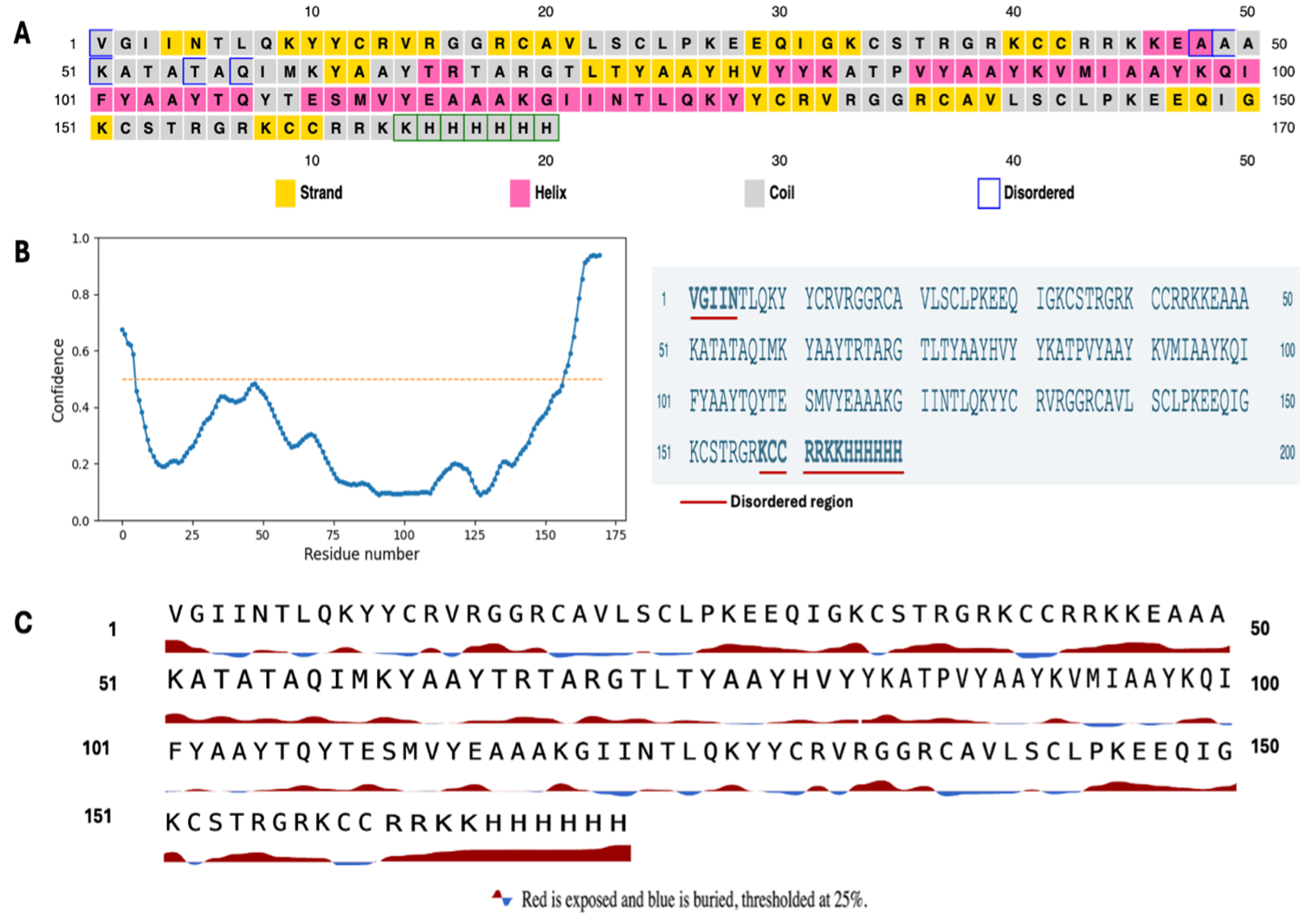
Predicted secondary structure features of the SPMEV vaccine construct. (A) Secondary structure composition showing α-helices (pink), β-strands (yellow), coils (gray), and disordered regions (blue). (B) Intrinsic disorder prediction indicating mostly ordered residues with limited flexibility at the C-terminal region. (C) Solvent accessibility plot demonstrating that the majority of CTL epitopes are surface-exposed, supporting their accessibility for antigen recognition
Disorder analysis indicated that most residues adopt structured conformations with only localized flexibility near the terminal polyhistidine tag. Such intrinsic disorder is not detrimental; in fact, moderate disorder in vaccine constructs can enhance antigen processing by the proteasome and promote efficient MHC presentation. This aligns with immunological evidence that disordered or flexible peptide regions are more prone to proteolytic cleavage, thereby increasing the likelihood of epitope liberation and presentation to cytotoxic T lymphocytes (CTLs).38
Surface accessibility predictions further supported the immunogenic potential of the construct, as the majority of CTL epitopes were located in solvent-exposed regions. Epitope exposure at the protein surface is essential for recognition by antigen-presenting cells and subsequent T-cell priming.20 Meanwhile, the burial of hydrophobic residues within the protein core suggests proper folding and minimized aggregation propensity, which are critical for vaccine stability and manufacturability.
Tertiary Structure of the SpMEV Construct
The quality of the predicted three-dimensional protein structure was assessed before and after refinement using stereochemical and energetic validation tools (Figure 5). The initial Ramachandran plot analysis revealed that 92.9% of residues were located in favored regions, with a minor fraction occupying disallowed conformational space. Following successive refinements using ModRefiner from the I-TASSER platform and GalaxyRefine2 from the GalaxyWeb server, the stereochemical accuracy improved substantially, yielding 98.7% of residues within the favored regions and, notably, a complete absence of residues in disallowed regions. This marked enhancement reflects the successful correction of backbone torsional strain and indicates a more realistic accommodation of secondary structure elements within energetically permissible conformations.39
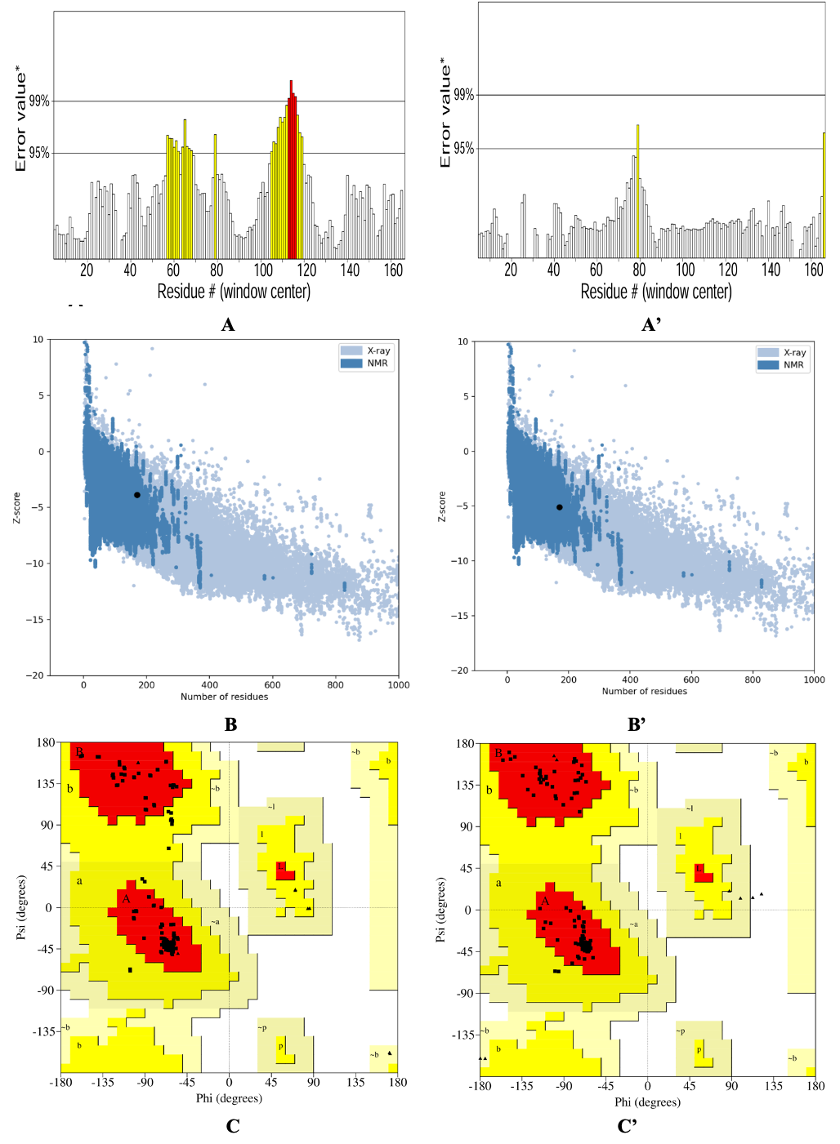
Initial (A to C) and refined (A’ to C’) three-dimensional structure of the SPMEV vaccine construct. The final model, refined using ModRefiner (I-TASSER) and GalaxyRefine2 (GalaxyWeb), demonstrates improved stereochemical and energetic quality. Structural domains including the adjuvant, CTL epitopes, linkers, terminal valine, and polyhistidine tag are indicated. The construct exhibits a stable, native-like conformation, ensuring epitope accessibility for antigen processing and presentation
Consistent with these improvements, the ERRAT quality factor increased from 82.69% in the unrefined model to 98.60% after refinement. Since ERRAT evaluates the statistics of non-bonded interactions, this elevation signifies a substantial reduction in steric clashes and non-ideal contacts, leading to improved atomic packing and greater structural stability.40 Such a refinement outcome is characteristic of the effective optimization of hydrogen bonding networks and hydrophobic core interactions, both of which are crucial for stabilizing protein folds.
The improvement was further supported by ProSA analysis, where the z-score shifted from -3.87 to -5.23. As ProSA z-scores benchmark protein models against experimentally resolved structures of comparable size, the refined structure’s more negative value suggests a closer approximation to a native-like fold with improved thermodynamic plausibility.41 Importantly, the convergence of these independent validation metrics strongly indicates that the applied refinement protocols were not only effective in correcting local stereochemical errors but also in enhancing the global energetics of the protein model.
The combined use of ModRefiner and GalaxyRefine2 likely underpins the observed improvements, as each tool contributes distinct refinements: ModRefiner emphasizes atomic-level geometry and torsional accuracy, while GalaxyRefine2 performs iterative side-chain repacking and relaxation of the entire structure.42 Thus, these refinements yielded a model with structural quality metrics approaching those of experimentally determined proteins. The resulting structure, free from disallowed torsions and exhibiting high-quality packing statistics, provides a more reliable basis for downstream functional annotation, docking simulations, or dynamic studies. The refined three-dimensional construct of the designed SpMEV with labels is shown in Figure 6.
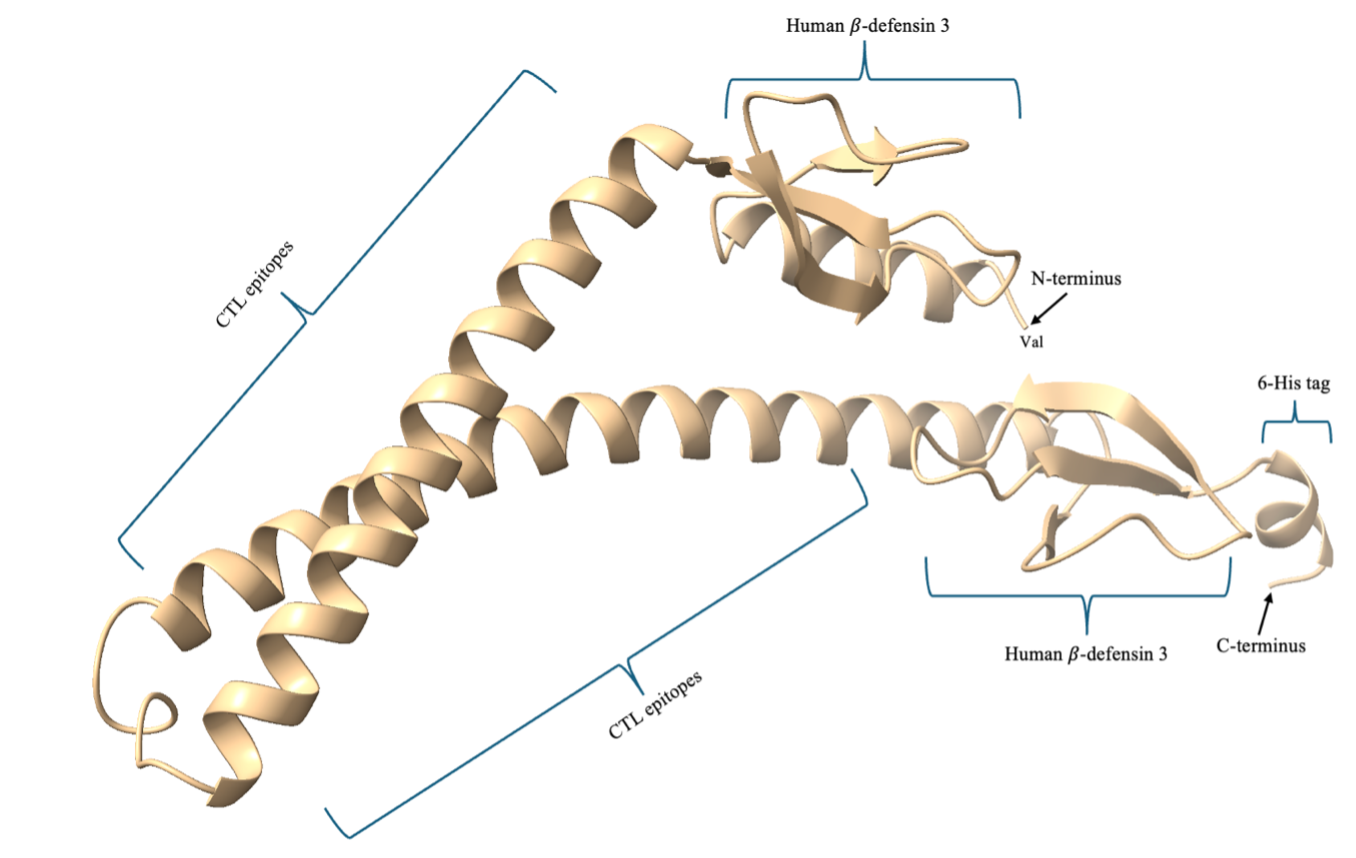
Refined three-dimensional structure of the SPMEV vaccine construct
Discussion
Determinants of Epitope Selection and Immunological Relevance
Notably, the number of predicted CTL epitopes varied among the selected S. pyogenes exoenzymes, with streptolysin O (SLO) yielding a higher epitope count compared to the other proteins. While a greater epitope yield may contribute to broader HLA population coverage, epitope abundance alone does not necessarily correlate with immunodominance or in vivo immunogenicity. Antigen processing efficiency, proteasomal cleavage, TAP transport, MHC class I binding stability, and T-cell receptor recognition collectively influence the immunological relevance of individual epitopes. Therefore, the differential epitope distribution observed across exoenzymes should be interpreted as a predictive indicator rather than a definitive measure of vaccine efficacy.
The cumulative filtering process substantially reduced the dataset but improved its overall quality, yielding epitopes that are non-allergenic, non-toxic, and non-cross-reactive. This stringent multi-tiered screening ensures that only biologically safe and immunologically relevant peptides are advanced for downstream analyses, thereby enhancing their translational feasibility for vaccine development. Moreover, this rigorous selection framework strengthens the specificity, safety, and immunological fidelity of the vaccine construct, minimizing the risk of adverse immune reactions and increasing its potential for regulatory approval and clinical success.
Implications of HLA Binding Promiscuity and Population Coverage in Multi-Epitope Vaccine Design
A central challenge in epitope-based vaccine design is overcoming the extensive polymorphism of HLA class I molecules, which differ substantially across global populations.26 Because cytotoxic T lymphocytes (CTLs) recognize antigens only when presented by compatible HLA alleles, epitope selection must prioritize promiscuous peptides capable of binding multiple alleles to achieve broad immunogenic coverage. Previous analyses indicate that targeting a subset of such promiscuous epitopes can yield over 90% global population coverage, although additional allele representation remains critical in genetically diverse regions such as Africa and Southeast Asia.43
Compared with recent in silico multi-epitope vaccine design against S. pyogenes43, the present work introduces three notable advances. First, the dominant inclusion of SLO-derived epitopes confers markedly higher global coverage (>90%) than earlier M-protein–focused constructs. Second, the downstream incorporation of β-defensin-3 as an intrinsic adjuvant is designed to enhance both mucosal and cellular immune activation, addressing limitations in prior models that lacked endogenous adjuvanticity. Third, the integration of regional HLA allele frequency analysis, particularly highlighting Philippine-prevalent alleles (HLA-A11:01, HLA-B*15:02), provides a geographically nuanced understanding often absent in global GAS vaccine pipelines.
The strong global population coverage of SLO epitopes underscores their value as universal vaccine components, consistent with their high conservation and immunodominance as cholesterol-dependent cytolysins eliciting potent CD8⁺ T-cell responses.2 Similarly, SpeB and DNase epitopes demonstrated broad multi-allelic binding capacity (78.00% and 77.02%), reflecting their conserved and surface-accessible nature, which favors efficient MHC class I presentation.17 Although MF and NADase achieved slightly lower coverage (71.72% and 70.39%), both exceed the 70% global feasibility threshold.44 MF’s modest coverage likely reflects its lower epitope yield, whereas NADase contributes epitopes essential for intracellular immune modulation and synergistic function with SLO.19 These findings demonstrate that incorporating SLO as the dominant epitope source, alongside complementary antigens such as SpeB, DNase, MF, and NADase, yields a multi-antigen, multi-epitope framework capable of achieving both global and region-specific population coverage.45
Structural and Thermodynamic Validation of Epitope–HLA Interactions
Molecular docking provides structural and energetic validation of peptide–HLA interactions, enabling prediction of binding affinity, orientation, and stability.46 While docking offers valuable insights into epitope–HLA binding behavior, it represents a static snapshot of molecular interactions. To fully assess the stability, flexibility, and dynamic behavior of these complexes under physiological conditions, molecular dynamics simulations would be necessary in future studies.
Thermodynamic principles further corroborate the docking results, as spontaneous binding occurs when ΔG < 0, with more negative values indicating stronger association. Correspondingly, the low K values observed reflect tight ligand–receptor binding and minimal complex dissociation, hallmarks of immunogenic stability. The consistency between ΔG, K, and docking conformations confirms that the selected epitopes form energetically favorable, structurally stable, and TCR-compatible complexes with their HLA alleles.
Hence, SLO-, SpeB-, and NADase-derived epitopes demonstrated the most stable and energetically favorable docking profiles, aligning with their previously observed high immunodominance and population coverage. It is important to note that the ΔG values predicted by the PRODIGY server have an estimated uncertainty of ±1.9 kcal/mol (RMSE), reflecting inherent model variability. Consequently, small differences in docking scores between epitopes should be interpreted cautiously, as they may fall within the prediction error range rather than indicating true differences in binding strength. Nonetheless, the overall trend of consistently low ΔG and K values across the selected complexes supports their strong binding potential and biological relevance. Collectively, these structural and thermodynamic validations confirm that the selected epitopes possess robust MHC class I binding, high structural fidelity, and strong immunogenic potential, underscoring their suitability for rational design of a multi-epitope vaccine against Streptococcus pyogenes.
Design Rationale, Safety Considerations, and Translational Implications of the SpMEV Construct
While humoral coverage through helper T-cell (HTL) and B-cell epitopes is important for achieving broad protection, this study specifically optimized the cytotoxic arm, with inclusion of HTL/B-cell epitopes planned in future construct iterations. The deliberate focus on CTL activation reflects the need to target intracellular persistence mechanisms of S. pyogenes, where cytotoxic immunity plays a central role in pathogen clearance.
Given the dual immunomodulatory nature of human β-defensin-3, several precautions were adopted to mitigate potential autoreactive or adverse effects. The full-length human hBD-3 sequence was employed to avoid neo-epitope formation, and all vaccine peptides were rigorously screened against the human proteome to exclude potential autoreactive epitopes. Additionally, computational docking and epitope mapping were performed with the understanding that in silico predictions cannot fully capture immunogenic variability, cytokine milieu effects, or host-specific responses, which may influence the overall adjuvant activity.
Future experimental validation should include comprehensive safety and efficacy assessments. These may encompass dose-escalation studies in murine models, profiling of Th1/Th2/Th17/Treg cytokines, autoantibody screening, histopathological examinations, and clinical monitoring for autoimmune manifestations. Importantly, these studies would clarify whether hBD-3 functions reliably as a safe endogenous adjuvant across different tissues and hosts or if alternative adjuvants with more predictable immunostimulatory profiles should be considered. Until such empirical evidence is available, any translational claims regarding hBD-3 use in humans should be interpreted with caution.
Immunogenicity and Safety Implications of the SpMEV In Silico Profile
The in silico evaluation of antigenicity, allergenicity, toxicity, and cross-reactivity provides essential preliminary validation of the SpMEV construct prior to experimental testing. The high antigenicity scores obtained from both VaxiJen and ANTIGENpro indicate strong intrinsic immunogenic potential, which is a critical requirement for effective vaccine design. These findings support the rationale that the selected epitope combination can elicit a robust immune response upon administration.
The absence of predicted allergenicity and toxicity further reinforces the biosafety of the construct. The low Tanimoto similarity index and negative AllerCatPro prediction suggest minimal risk of hypersensitivity or off-target immune activation, which is a common limitation in peptide-based vaccines. Additionally, the lack of significant similarity with human proteins reduces the likelihood of autoimmune cross-reactivity, an important consideration for translational vaccine development.
Together, these results demonstrate that the SpMEV construct satisfies key immunological and safety benchmarks required for further development. While computational predictions provide valuable insight, experimental validation remains essential to confirm these findings under physiological conditions. Nonetheless, the favorable in silico profile supports progression of the SpMEV construct toward structural validation, in vitro immunogenicity assays, and subsequent in vivo evaluation.
Structural Features and Implications for Vaccine Design
The secondary structure composition, limited intrinsic disorder, and favorable solvent accessibility profile indicate that the SPMEV vaccine construct is structurally stable yet sufficiently flexible to allow efficient epitope processing and presentation. These features are consistent with effective immunogen design principles, supporting the construct’s potential to elicit robust and targeted immune responses.20,38
The balance between structural rigidity and flexibility is particularly important in multi-epitope vaccines, where excessive rigidity can hinder antigen processing, while excessive disorder may compromise structural integrity. In this construct, the predominance of ordered secondary structural elements ensures conformational stability, while localized disordered regions may facilitate proteasomal cleavage and MHC loading. This structural configuration aligns well with current understanding of antigen processing pathways and reinforces the suitability of the SpMEV construct for further experimental validation.
The observed improvements in stereochemical accuracy, atomic packing, and global energetics strongly support the structural reliability of the SpMEV construct. The increase in residues within favored Ramachandran regions, elevated ERRAT quality factor, and more negative ProSA z-score collectively indicate a model approaching native-like conformations. These structural enhancements are critical for multi-epitope vaccine design, as accurate three-dimensional folding ensures proper epitope orientation, stability, and accessibility for immune recognition.39
The refinement protocols applied—ModRefiner for atomic-level geometry and GalaxyRefine2 for side-chain repacking and relaxation—demonstrate the importance of combining complementary computational approaches to achieve high-quality protein models. A structurally robust tertiary fold is essential not only for stability during storage and delivery but also for reliable downstream applications such as molecular docking, dynamic simulations, and experimental expression in vitro and in vivo.
Although these findings are based on computational predictions, the refined secondary and tertiary structures provide a strong foundation for further experimental validation, including molecular dynamics simulations, recombinant expression, and functional immunogenicity assessments. Ultimately, these high-quality structural features support the translational potential of the SpMEV construct as a rationally designed vaccine candidate.
Conclusion
This study established a comprehensive immunoinformatics framework to identify and prioritize conserved CTL epitopes from five major Streptococcus pyogenes exoenzymes. The refined, non-allergenic, and non-toxic epitopes showed strong HLA affinity and broad population coverage, with SLO and SpeB epitopes as top candidates. These epitopes were assembled into a rationally designed multi-epitope construct incorporating AAY linkers and human β-defensin-3 as an adjuvant to enhance immunogenic potential. In silico structural modeling and molecular docking analyses indicated that the construct possesses favorable stability, folding, and receptor-binding properties suggestive of its ability to elicit CD8⁺ T-cell responses. While these computational findings are encouraging, they require further validation through in vitro and in vivo experiments to confirm immunogenicity and safety prior to any translational application.
Future Direction
For the advancement of an epitope-driven vaccine against S. pyogenes, future studies should focus on the actual experimental evaluation of the predicted cytotoxic T lymphocyte (CTL) epitopes. Specifically, laboratory assays to confirm the epitope’s ability to bind MHC class I molecules, activate CD8⁺ T-cells, and induce cytokine responses including the release of interferon-gamma (IFN-γ), an important measure for cellular immunity. In vivo studies using HLA-transgenic mice and non-human primates, along with in vitro studies, are essential to validating immunogenicity, safety, dose optimization, and maintaining immune memory in a physiological context.
To broaden immune protection, future vaccine designs must also incorporate helper T-cell (HTL) and B-cell epitopes alongside CTL targets. This multi-epitope process will promote a more diverse immune response with cytokine support and the formation of antibodies. Advanced delivery systems for vaccines, including nanoparticle systems, liposomal delivery, or viral vectors may further boost antigen presentation and vaccine efficacy. Considering the differences in HLA allele frequencies in populations, especially in Southeast Asia, it is recommended to consider a region-centered target for specific epitope modification to maximize coverage. Continuous genomic surveillance of circulating S. pyogenes strains will also be important for monitoring any antigenic changes and maintaining prolonged vaccine effectiveness.
In addition, the feasibility of heterologous expression and formulation of the proposed multi-epitope construct warrants careful experimental evaluation. The inclusion of multiple CTL epitopes linked by AAY spacers and fused to an immunomodulatory adjuvant may influence protein folding, solubility, and stability across different expression systems. Factors such as codon optimization, host-specific post-translational processing, and the potential for aggregation should be systematically assessed during construct optimization. Addressing these considerations will be essential for translating the computationally designed vaccine candidate into a viable experimental and clinical formulation.
Abbreviations
ARF: acute rheumatic fever; CFTR: cystic fibrosis transmembrane conductance regulator; CTL: cytotoxic T-lymphocyte; DNase: streptodornase; GAS: Group A Streptococcus; GRAVY: grand average of hydropathicity; hBD-3: human β-defensin 3; HLA: human leukocyte antigen; HTL: helper T lymphocyte; IC₅₀: half-maximal inhibitory concentration; IEDB: Immune Epitope Database; IFN-γ: interferon-gamma; K D: dissociation constant; LMICs: low- and middle-income countries; MF: mitogenic factor; MHC: major histocompatibility complex; MSA: multiple sequence alignment; NADase: NAD-glycohydrolase; NETs: neutrophil extracellular traps; pI: isoelectric point; pMHC: peptide–MHC; ProSA: Protein Structure Analysis; PTM: post-translational modification; PVS: Protein Variability Server; RefSeq: Reference Sequence; RHD: rheumatic heart disease; RMSD: root-mean-square deviation; RMSE: root-mean-square error; SLO: streptolysin O; SpeB: streptococcal pyrogenic exotoxin B; SpMEV: Streptococcus pyogenes multi-epitope vaccine; TAP: transporter associated with antigen processing; TCR: T-cell receptor; TLR: Toll-like receptor; VFDB: Virulence Factor Database; ΔG: Gibbs free energy of binding
Acknowledgments
None.
Author’s contributions
All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have not used generative AI (a type of artificial intelligence technology that can produce various types of content including text, imagery, audio and synthetic data).
Competing interests
The authors declare that they have no competing interests.