

Harnessing genomic profiling for precision breast cancer therapies: Advances in HER2-targeted treatments, PARP inhibitors for BRCA mutations, and insights from clinical trials
- Centre for Applied Molecular Biology (CAMB), University of the Punjab, Quaid-e-Azam Campus, Lahore, Pakistan
- School of Biochemistry and Biotechnology (SBB), University of the Punjab, Quaid-e-Azam Campus, Lahore, Pakistan
- University Institute of Medical Lab Technology, Faculty of Allied Health Sciences. The University of Lahore -54590 Pakistan
Abstract
The therapeutic landscape for breast cancer has evolved considerably over time; specifically, the integration of genomic profiling and molecular genetics has improved personalized therapeutic strategies. Genomic profiling identifies mutations and allelic variations in breast cancer, enabling more targeted interventions. Genetic research has highlighted the overexpression or dysfunction of the HER2 receptor, ALKBH5 enzyme, BRCA proteins, and PARP enzymes in advanced breast carcinomas. Current treatment modalities include HER2-targeted agents and PARP inhibitors. The application of monoclonal antibodies (mAbs), tyrosine kinase inhibitors (TKIs), and antibody-drug conjugates (ADCs) to target overexpressed HER2 receptors in oncology patients has improved survival outcomes and treatment responses. In addition, ADCs have shown remarkable efficacy by selectively targeting cancer cells while sparing healthy tissue, utilizing target-specific monoclonal antibodies conjugated to cytotoxic payloads. Conversely, PARP inhibitors are primarily indicated for triple-negative breast cancer (TNBC) cases due to the high prevalence of BRCA1/BRCA2 mutations that impair DNA repair mechanisms. This review provides a comprehensive analysis of breast cancer treatments and associated clinical trials, including genomic profiling and drugs targeting the HER2 receptor [mAbs, ADCs, and TKIs] and PARP inhibitors. Furthermore, we examine the effect of these therapeutic agents on BRCA1/2 mutations and other relevant enzymatic pathways. Despite these advancements, certain challenges persist, such as the emergence of drug resistance, underscoring the need for further research and innovative techniques to overcome these limitations.
Introduction
Integrating genomic profiling and targeted therapies in breast carcinoma
In many nations, there is significant concern regarding the rising incidence of breast malignancies among the female population 1. According to the World Health Organization (WHO), in 2022, breast cancer accounted for approximately 2.3 million cases globally, representing 11.6 percent of all reported cancer incidences (). Consequently, it is now recognized as the most pervasive type of cancer across both genders in 156 out of the 185 countries analyzed 2. In addition, it remains one of the most lethal malignancies and was estimated to have caused around 670,000 deaths in 2022; thus, it is the leading cause of cancer-related mortality, particularly for women worldwide 3. While various diagnostic modalities and therapeutic interventions have been developed, the inherent biological complexity of the disease remains a formidable challenge.
Heterogeneity of breast tumors
Breast cancer is not a single pathological entity but rather a highly heterogeneous disease; this realization represents one of the most critical advancements in the field of oncology 4. It comprises a spectrum of biologically heterogeneous subtypes defined by distinct molecular and genetic profiles. Such insights have fostered the development of precision oncology, which focuses on the unique molecular signatures of a patient’s malignancy. At the heart of this approach is genomic characterization, which has transformed the understanding and management of breast cancer by facilitating the identification and implementation of personalized therapeutic regimens 4,5.
For extended periods, breast cancer management relied upon generalized therapeutic strategies, including chemotherapy, radiation, and hormonal therapies 6,7. Although these modalities have saved many lives, they do not account for the genetic and molecular heterogeneity inherent in the tumor. Therefore, their efficacy is patient-specific, and patients may experience significant adverse effects with marginal clinical improvement 8. In response to this therapeutic gap, genomic profiling has been developed to elucidate the precise genetic profile of each tumor. This technique analyzes gene amplification, somatic mutations, and other alterations in the molecular landscape, including the DNA and RNA of tumor cells 9.
Literature search methodology and risk-of-bias assessment
To enhance transparency and reproducibility, this review was conducted using a systematic literature-search strategy adhering to the PRISMA guidelines for scoping reviews. Three major electronic databases—PubMed/MEDLINE, Scopus, and Web of Science—were comprehensively searched up to June 30, 2024. The search strategy was developed to capture both preclinical and clinical studies relevant to breast cancer therapeutics, involving HER2-targeted monoclonal antibodies (mAbs), tyrosine kinase inhibitors (TKIs), antibody-drug conjugates (ADCs), BRCA mutations, PARP inhibitors, and RNA-modifying enzymes including ALKBH5, METTL3, and FTO. Keywords and Medical Subject Headings (MeSH) terms were combined using Boolean operators: ("HER2" OR "ERBB2") AND ("breast cancer") AND ("antibody drug conjugate" OR "trastuzumab" OR "pertuzumab" OR "tyrosine kinase inhibitor") AND ("resistance" OR "clinical trial"); and ("BRCA1" OR "BRCA2") AND ("breast cancer") AND ("PARP inhibitors" OR "olaparib" OR "talazoparib" OR "niraparib"). For RNA-modifying enzymes, the search included terms such as ("ALKBH5" OR "METTL3" OR "FTO") AND ("m6A modification") AND ("breast cancer"). Reference lists of pivotal articles and reviews were manually searched to ensure comprehensive coverage.
Inclusion criteria were: (i) original peer-reviewed articles, systematic reviews, or meta-analyses; (ii) clinical trials (phases I–III) and major observational studies reporting therapeutic efficacy, resistance mechanisms, or genomic profiling insights in breast cancer; and (iii) mechanistic preclinical studies utilizing cell lines or animal models that addressed HER2-targeted therapy, PARP inhibitors, or RNA-modifying enzymes. Only studies published between January 2015 and June 2024 and written in English were included. Exclusion criteria comprised: (i) case reports, conference abstracts, and unpublished theses; (ii) studies with insufficient or anecdotal data; and (iii) reports lacking direct relevance to breast cancer or genomic-targeted therapy.
The final dataset consisted of approximately 90 publications, including 22 clinical trials (comprising randomized controlled trials and Phase II/III studies), 10 systematic reviews/meta-analyses, and over 50 mechanistic/preclinical studies. Clinical trial identifiers (NCT numbers) were extracted and are presented in the relevant tables to ensure traceability.
Methodological quality and risk-of-bias assessments were conducted. For clinical trials, methodological rigor was evaluated by examining trial phase, randomization protocols, blinding procedures, sample size, and the completeness of outcome reporting. Trials with limited cohort numbers, a lack of blinding, or incomplete reporting were identified as carrying a higher risk of bias. For preclinical studies, we considered experimental reproducibility, the use of appropriate controls, and independent validation. While the heterogeneity of the included studies precluded a universal scoring system, the Cochrane Risk-of-Bias 2 (RoB 2.0) tool was applied where appropriate to evaluate five domains: (i) the randomization process, (ii) deviations from planned interventions, (iii) missing outcome data, (iv) measurement of the outcomes, and (v) selection of the reported results. These domains were rated as "low risk," "some concerns," or "high risk."
An overview of ten prominent randomized trials involving HER2-targeted and PARP-inhibitor therapies is provided in Table X, facilitating a rapid assessment of the available level of evidence. The majority of Phase III studies (e.g., CLEOPATRA, DESTINY-Breast03, OlympiAD) utilizing large-scale, multicenter designs demonstrated robust randomization and objective endpoints with minimal attrition bias. However, some open-label designs (e.g., SOPHIA, BROCADE3) presented a moderate risk of performance bias.
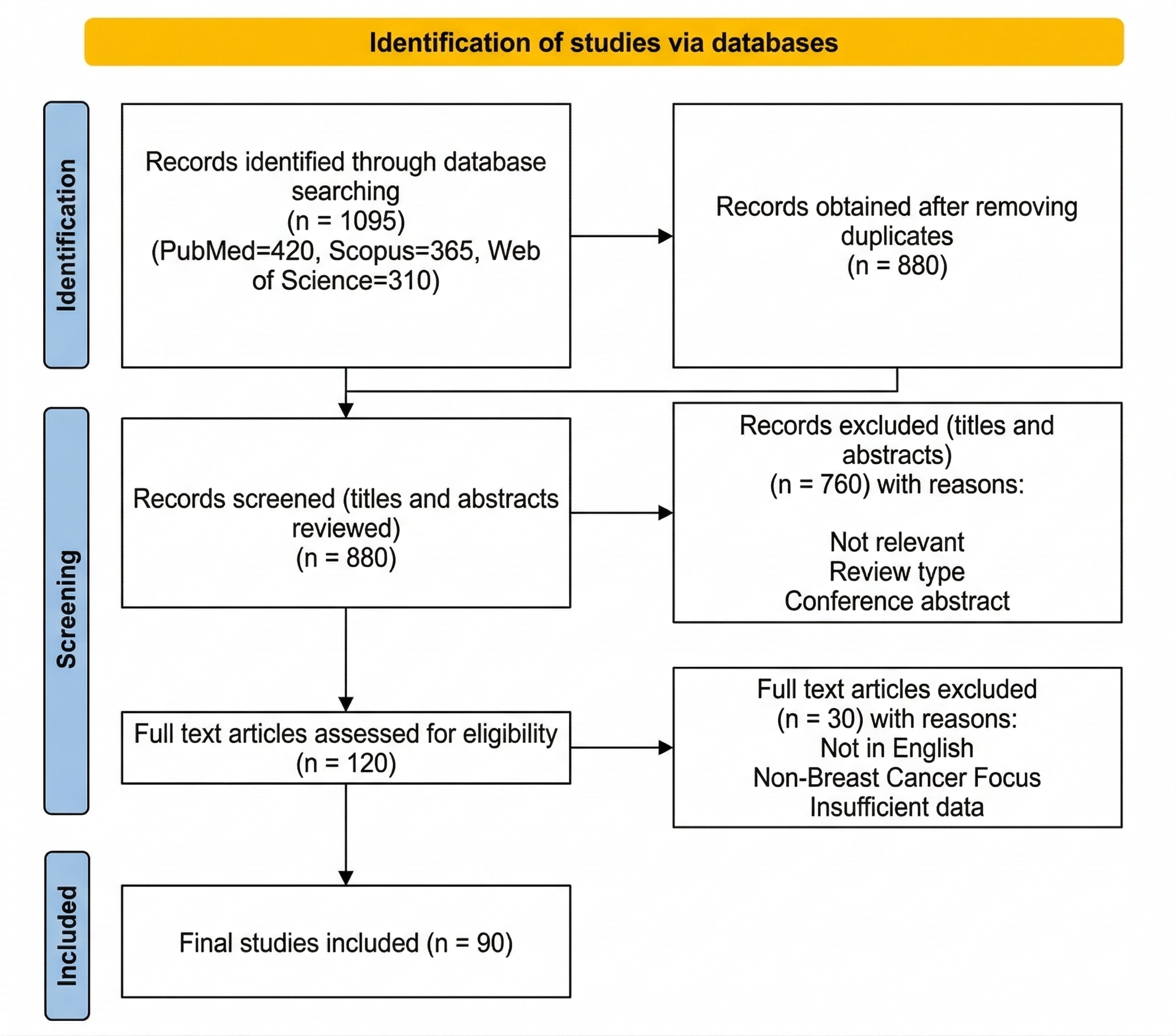
PRISMA 2020 Flow Diagram of Study Selection. This diagram illustrates the systematic literature search and screening process conducted across three major electronic databases: PubMed ($n = 420$), Scopus (n = 365), and Web of Science (n = 310). After the removal of 215 duplicates, 880 records were screened by title and abstract, resulting in the exclusion of 760 entries categorized as irrelevant, conference abstracts, or non-peer-reviewed types. A total of 120 full-text articles were assessed for eligibility, of which 30 were excluded due to being in languages other than English, lacking a breast cancer focus, or providing insufficient data. The final inclusion consists of 90 studies, including 22 clinical trials and over 50 mechanistic studies, evaluating genomic profiling, HER2-targeted therapies, and PARP inhibitors in breast cancer.
Comprehensive risk-of-bias assessment for pivotal randomized controlled trials evaluating HER2-targeted and BRCA/PARP-directed breast cancer therapies. This table summarizes the methodological quality of ten landmark clinical trials (2015–2022), including CLEOPATRA, DESTINY-Breast03, and OlympiAD. Each study is evaluated across five domains: randomization process, blinding protocols, outcome reporting accuracy, management of missing data, and overall risk of bias. The analysis highlights the shift from double-blind designs to open-label trials with objective endpoints, such as Progression-Free Survival (PFS) and Overall Survival (OS), providing a snapshot of the current level of evidence for precision oncology interventions.
| Study (Year) | Intervention | Randomization | Blinding | Outcome Reporting | Missing Data | Overall Risk | Key Remarks |
|---|---|---|---|---|---|---|---|
| CLEOPATRA (2015) | Pertuzumab + Trastuzumab + Docetaxel vs placebo | Low | Double-blind | Low | Low | Low | Large multicentre phase III trial; clear OS benefit |
| DESTINY-Breast 03 (2021) | T-DXd vs T-DM1 | Low | Open-label | Low | Low | Low | objective endpoints (PFS/OS); independent review |
| DESTINY-Breast 04 (2022) | T-DXd vs chemo in HER2-low | Low | Open-label | Low | Some | Low–Moderate | Robust PFS gain; ILD toxicity noted |
| SOPHIA (2019) | Margetuximab vs Trastuzumab + Chemo | Some concern | Open-label | Low | Low | Moderate | Performance bias possible |
| EMBRACA (2018) | Talazoparib vs Standard Chemo | Low | Open-label | Low | Low | Low | Clear PFS advantage; hematologic AEs |
| OlympiAD (2017) | Olaparib vs Standard Chemo | Low | Open-label | Low | Low | Low | Adequate allocation; independent assessment |
| BROCADE3 (2020) | Rucaparib + Chemo vs Chemo | Low | Double-blind | Low | Low | Low | Randomized; consistent reporting |
| NOVA (2016) | Niraparib vs Placebo in HR-def ovarian/breast subset | Low | Double-blind | Low | Low | Low | Relevant safety data for breast subset |
| ARIEL3 (2017) | Rucaparib vs Placebo | Low | Double-blind | Low | Low | Low | Clear statistical rigor and PFS gain |
| I-SPY 2 (2020) | Veliparib + Carboplatin + Chemo vs Chemo alone | Low | Randomized adaptive | Some | Some | Moderate | Adaptive design; high innovation value |
Genomic Profiling
According to Davis and Zhang 10, the development of advanced genomic techniques has enabled the mapping of tumors with extraordinary accuracy. Modalities such as next-generation sequencing (NGS) enable the assessment of genetic alterations across the entire genome of a tumor, facilitating the identification of distinct oncogenic drivers that are amenable to targeted therapy.
Furthermore, liquid biopsies represent a non-invasive alternative to traditional tissue biopsies; these include the analysis of circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA). This method not only detects genetic mutations but also monitors clonal evolution over time, thereby providing real-time insights into therapeutic efficacy or the emergence of resistance.
Additionally, multigene expression assays, such as Oncotype DX and MammaPrint, provide further clinical utility by quantifying the risk of recurrence, thereby guiding the administration of adjuvant therapies in early-stage breast cancer 11.
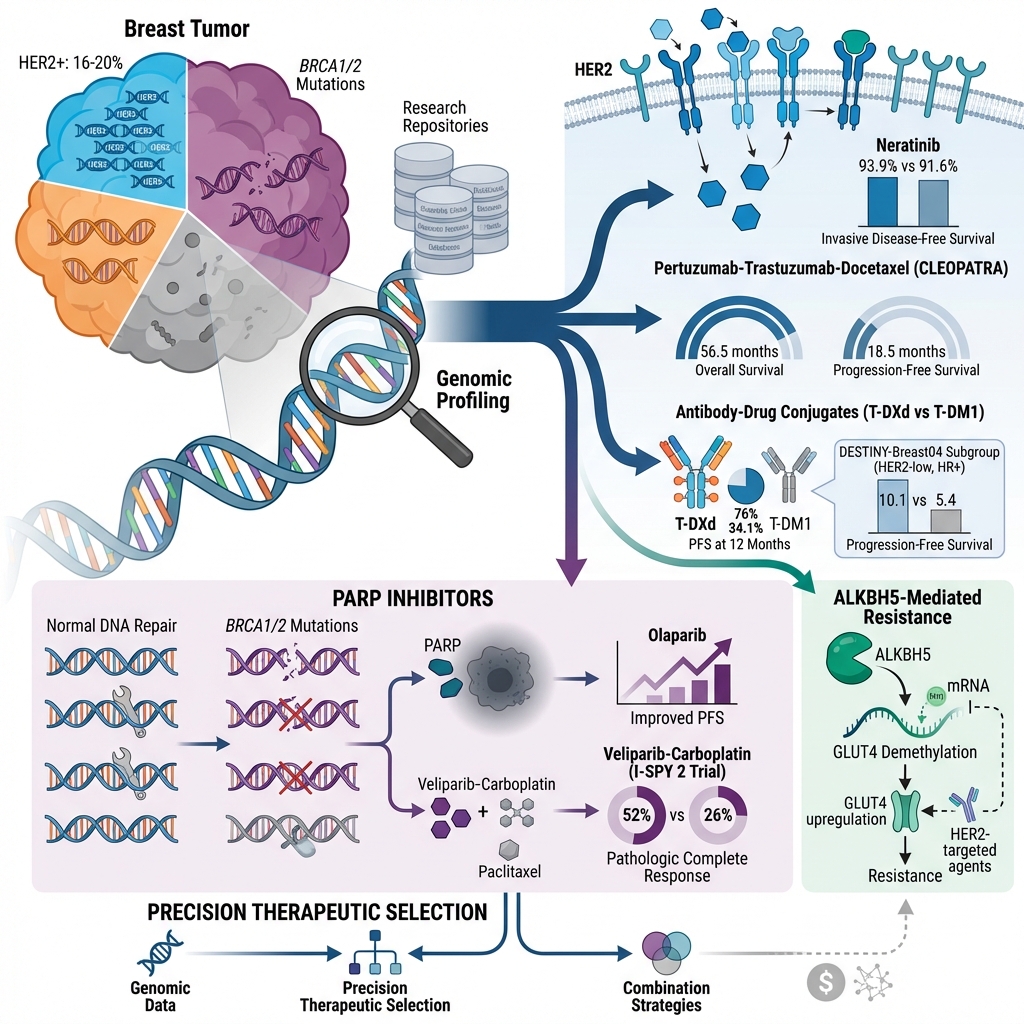
Novel alterations revealed by genomic profiling include multiple actionable genetic changes that have transformed the therapeutic landscape of breast cancer. The most prominent genetic alteration is the overexpression of the HER2 gene, which is reported in 16–20% of breast cancer patients. While HER2 amplification promotes aggressive malignancy, the identification of this alteration has led to the development of agents targeting HER2, such as trastuzumab and pertuzumab 12,13. These therapeutics target the HER2 receptor, thereby halting tumor growth and greatly increasing survival rates among patients with HER2-positive breast cancer 14.
Likewise, genomic biomarkers have identified BRCA1/BRCA2 mutations as being of prime importance in triple-negative breast cancer (TNBC). These mutations impair DNA repair mechanisms, rendering cancer cells susceptible to PARP inhibitors, including olaparib and talazoparib 15. PARP inhibitors work by targeting specific hallmarks of cancer, notably the inability of tumor cells to repair double-strand DNA breaks through homologous recombination. By inhibiting PARP, cancer cells are selectively killed via synthetic lethality while normal tissue is spared 13.
Beyond HER2 and BRCA mutations, genomic studies have identified other targetable alterations. For example, PIK3CA mutations are frequently detected in hormone receptor-positive (HR+), HER2-negative breast tumors. These mutations promote tumorigenesis through the PI3K/AKT/mTOR signaling pathway and are managed with PI3K inhibitors such as alpelisib 16. Moreover, novel targets, including components of the AKT pathway and FGFR alterations, are being explored within clinical trials as emerging therapeutic targets 17,18.
According to Tirada and Aujero 19, genomic profiling has standardized clinical practice by improving patient outcomes. It permits a personalized approach that optimizes efficacy while reducing off-target toxicity. For instance, a patient with HER2-positive breast cancer may be prescribed trastuzumab, which has been shown to significantly improve survival compared to chemotherapy alone 20. When resistance to HER2-targeted therapy develops, genomic profiling can characterize secondary mutations in the PI3K/AKT signaling pathways, enabling combination treatments to overcome this resistance. Likewise, for a patient with TNBC and a BRCA1 mutation, PARP inhibitors may be utilized, and constant longitudinal monitoring with liquid biopsies allows for treatment modification in case of resistance 21.
The integration of genomic profiling into breast cancer care offers multiple advantages, although significant obstacles remain. One challenge is reducing the cost of genomic tests and targeted therapies, which can be prohibitively high, acting as a barrier to access for most patients, especially in developing countries 22,23. Furthermore, translating the vast volume of data produced by genomic analyses presents a challenge due to its complexity, requiring advanced bioinformatic tools. Other considerations include ethical concerns regarding informed consent and genomic data privacy 24.
Further studies are required to refine and broaden the application of genomic profiling. In order to obtain a more detailed picture of tumor biology, genomic, proteomic, and metabolomic data are being integrated via multi-omics analysis. Machine learning and artificial intelligence are also being employed in genomic analysis to predict treatment responses and identify novel targets 25. Approaches such as personalized cancer vaccines, tailored to individual neoantigens, are other promising treatment options. Also, advancements in liquid biopsy technology enable the tracking of clonal evolution for real-time modifications to treatment regimens 26.
In conclusion, identifying the signaling pathways and mutations that control tumor progression has led to the development of specific drugs that improve clinical outcomes tremendously with minimal side effects. As exemplified by HER2-targeted agents and PARP inhibitors, the integration of genomic data into clinical practice is the primary tenet underlying precision oncology. However, ensuring treatment accessibility, affordability, and the robust interpretation of complex data remains paramount. As research advances and the feasibility of comprehensive genomic profiling increases, the prospect of individualized breast cancer treatment becomes a reality for millions of patients worldwide 25,27.
HER2 Targeted treatment for breast carcinoma
The Human Epidermal Growth Factor Receptor 2 (HER2) receptor, also known as ErbB2, serves as a significant biomarker in breast cancer; it is vital for both diagnosis and the development of targeted therapeutic strategies 28. In the majority of cases, HER2 overexpression is observed in breast carcinoma patients, who are commonly treated with trastuzumab and chemotherapy 29. Before 2020, lapatinib was the only FDA-approved tyrosine kinase inhibitor (TKI) used against HER2-positive breast carcinoma. While monoclonal antibodies are also available, their high molecular weight prevents them from crossing the blood-brain barrier (Table 1) 30. Consequently, oral HER2-targeted kinase inhibitors—including tucatinib, neratinib, and pyrotinib—are employed to prevent Central Nervous System (CNS) metastasis in patients diagnosed with HER2-positive carcinomas 31.
Monoclonal antibodies and tyrosine kinase inhibitors overview
A multicenter, randomized, double-blind, placebo-controlled, Phase III trial was conducted at 495 research centers across North America, Europe, Asia, and South America to evaluate the efficacy of neratinib—an irreversible tyrosine kinase inhibitor (TKI) targeting HER1, HER2, and HER4—in patients with malignant breast carcinoma. The study enrolled 2,840 women aged 18 years or older with stage I–III HER2-positive breast cancer who had previously completed chemotherapy and trastuzumab-based adjuvant therapy. Participants were randomly assigned to receive either neratinib (240 mg/day) or a placebo for one year.
Two-year follow-up data demonstrated that neratinib significantly improved invasive disease-free survival compared to the placebo group. The cancer-free survival rate was 93.9% in the neratinib group versus 91.6% in the placebo group, representing a 33% reduction in the risk of disease progression or death ($p = 0.0091$) 28. However, neratinib was associated with a higher incidence of adverse events, specifically diarrhea; therefore, proactive management and continued longitudinal follow-up are required for further clinical assessment. The biochemical mechanism of TKIs in HER2-positive breast cancers is illustrated in Figure 229.
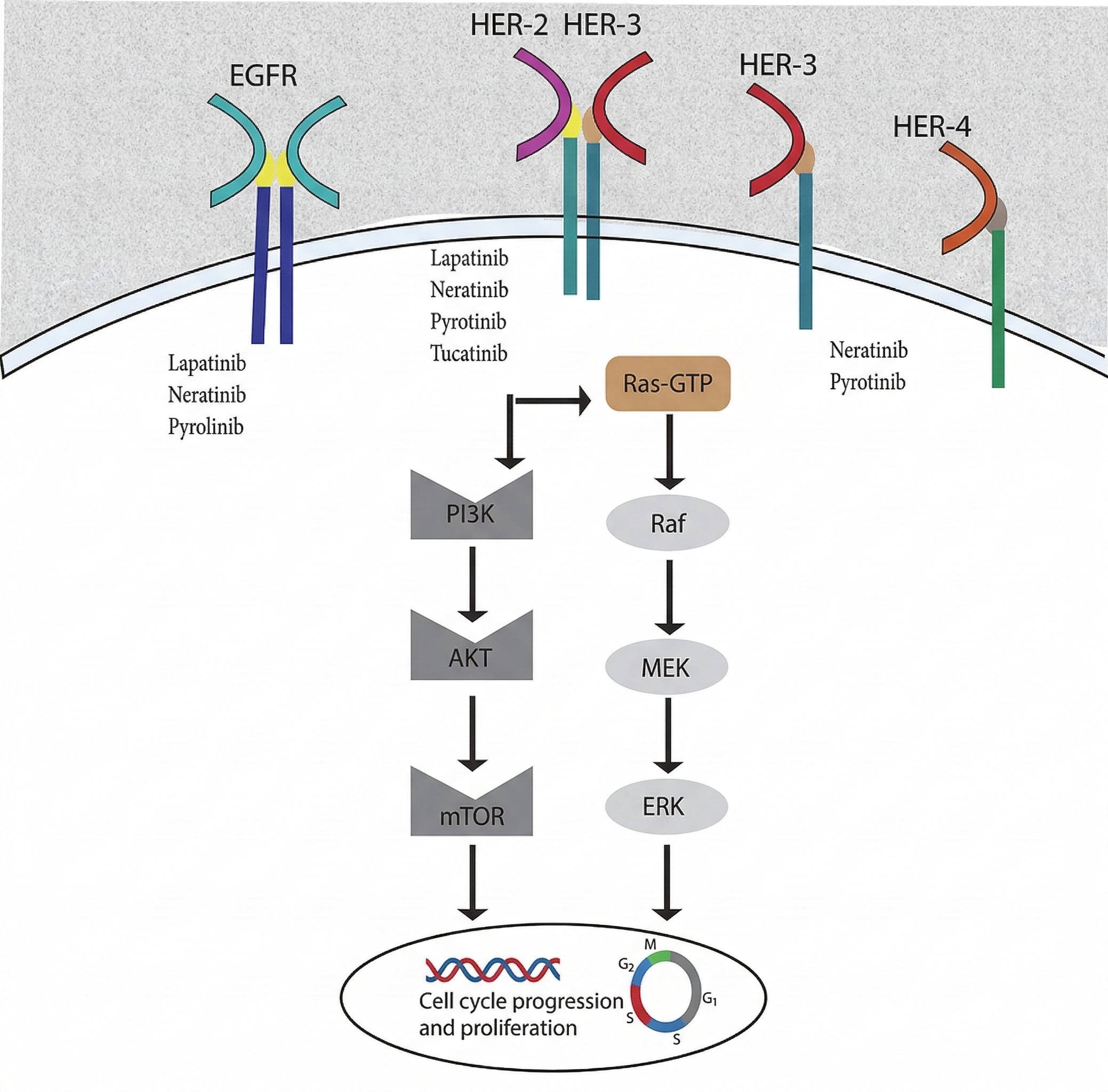
Schematic representation of the biochemical mechanisms and downstream signaling pathways targeted by HER2-selective Tyrosine Kinase Inhibitors (TKIs). This figure illustrates the inhibition of the Human Epidermal Growth Factor Receptor (HER) family, including EGFR (HER1), HER2, HER3, and HER4. Unlike other receptors, HER2 has no known active ligand and is activated through homo- or heterodimerization, which triggers the phosphorylation of cytoplasmic tyrosine residues. Small-molecule TKIs such as lapatinib, neratinib, pyrotinib, and tucatinib penetrate the cell membrane to bind the intracellular kinase domain, effectively blocking the Ras-GTP/Raf/MEK/ERK and PI3K/AKT/mTOR signaling cascades. The disruption of these pathways inhibits the oncogenic signals required for cell cycle progression and tumor cell proliferation.
In a subsequent phase 1b clinical study, tucatinib (an oral, highly potent HER2-selective tyrosine kinase inhibitor) was evaluated as a novel treatment in combination with ado-trastuzumab emtansine (T-DM1) in patients with advanced HER2+ breast cancer, regardless of the presence of brain metastases. The study established the recommended dosages of tucatinib when administered in combination with T-DM1. Borges et al. 30 enrolled 57 participants aged ≥18 years with HER2+ metastatic breast cancer. Prior to enrollment, patients had received previous treatment with trastuzumab, pertuzumab, and T-DM1. The results suggest that the recommended dose of tucatinib is 300 mg twice daily.
In the clinical study known as the CLEOPATRA trial, Swain et al. evaluated HER2+ metastatic breast cancer patients who had not previously received chemotherapy or targeted therapy for metastatic disease. Participants were randomized to receive pertuzumab in addition to trastuzumab and docetaxel. The results demonstrated a significant extension of overall survival (OS; p < 0.001) and progression-free survival (PFS) by approximately 56.5 months and 18.5 months, respectively 31.
Characteristics and clinical efficacy of novel monoclonal and bispecific antibodies for HER2-positive breast cancer. This table details the molecular targets and clinical milestones of emerging immunotherapy agents, including Margetuximab, ZW25, and PRS-343. It highlights key features such as Fc-region optimization for enhanced antibody-dependent cellular cytotoxicity (ADCC), dual-epitope binding to overcome resistance, and the recruitment of T-cells to the tumor microenvironment. The table includes National Clinical Trial (NCT) identifiers and summarizes critical endpoints such as Maximum Tolerated Dose (MTD), objective Response Rate (ORR), and Progression-Free Survival (PFS) from Phase I to Phase III investigations.
| Antibody | Type/Target | Key Features | Trial Identifiers | Endpoints | References |
|---|---|---|---|---|---|
| Marget-uximab | Chimeric monoclonal antibody targeting similar HER2 epitope as trastuzumab with Fc region optimization | Enhances ADCC via high-affinity binding to CD16a |
Phase 1 (NCT01148849) SOPHIA trial (phase III) | Phase 1: No MTD identified; safe at amount up to ≈0.018 g/kg | |
| ZW25 | Bispecific antibody directing HER-2-epitope ECD2 and ECD4 | Dual binding enhances HER2 inhibition | Phase ½ (NCT02892123) | Phase 1/2: MTD10 mg/kg per week/20 mg/kg per two weekly | |
| PRS-343 | Bispecific antibody directing HER-2 and CD137 (4-1BB) | Stimulates CD-137-positive T-cells |
Phase1 (NCT03330561) Combination with Atezolizumab (NCT03650348) | Phase 1: Evaluating single-agent efficacy |
Yi et al. 34 conducted a study to investigate HER2 mutations in metastatic breast cancer (MBC) and evaluate the efficacy of pyrotinib in patients harboring these mutations. The study enrolled 1,184 patients with invasive breast cancer to characterize mutation profiles using circulating tumor DNA (ctDNA) sequencing via a 1021-gene panel. ctDNA sequencing identified 105 patients (8.9%) with HER2 mutations, comprising both amplification-positive and amplification-negative cases. A significantly higher frequency of mutations was observed in patients with HER2 amplification compared to those with negative amplification (19.5% vs. 4.8%, p < 0.001). Furthermore, progression-free survival (PFS)—calculated from treatment initiation (trastuzumab or pyrotinib therapy) to the date of disease progression—was notably shorter in HER2-mutated patients relative to those with wild-type HER2. In the subsequent phase II study, pyrotinib demonstrated a 40% objective response rate in patients with non-amplified MBC harboring kinase domain mutations. Conversely, patients with concurrent HER2 amplification and extracellular domain mutations demonstrated resistance to pyrotinib therapy.
Collectively, HER2-targeted tyrosine kinase inhibitors (TKIs) demonstrate how variations in molecular selectivity, treatment settings, and clinical profiles affect tolerability and efficacy. Neratinib, an irreversible inhibitor, demonstrated a moderate and statistically significant improvement in two-year invasive disease-free survival when administered as adjuvant therapy following trastuzumab; however, it was associated with significant diarrhea due to off-target inhibition of EGFR and HER4. This broad receptor inhibition limits treatment adherence and necessitates proactive symptom management 35. In contrast, tucatinib—a highly selective HER2 inhibitor—exhibited superior tolerance and central nervous system (CNS) activity when combined with ado-trastuzumab emtansine, thereby providing clinical benefits to patients with brain metastases 36. Furthermore, pertuzumab, a monoclonal antibody that prevents HER2 dimerization at the extracellular domain, achieved the most significant improvement in overall survival and PFS when combined with trastuzumab and docetaxel in the CLEOPATRA trials 37. Simultaneously, pyrotinib showed promising efficacy in HER2-mutated cancers (specifically kinase domain mutations), though resistance remains a critical concern 34.
The major challenge in treating HER2-positive breast carcinoma is the development of resistance to standard-of-care agents such as trastuzumab and lapatinib. To address this, Liu et al. 38 investigated the role of the $m^6A$ demethylase enzyme ALKBH5. ALKBH5 expression is significantly upregulated in breast carcinoma, where it promotes cancer cell progression by stabilizing GLUT4 mRNA. By removing $m^6A$ modifications, ALKBH5 increases the stability of GLUT4 transcripts, leading to upregulated GLUT4 protein expression and enhanced glycolysis. Consequently, Liu demonstrated that targeting the ALKBH5/GLUT4 pathway through genetic knockdown or knockout may serve as a viable therapeutic strategy for HER2-positive breast cancer 39.
Antibody Drug Conjugates (ADC) against HER-2 +ve cancers
In addition to these treatments, antibody-drug conjugates (ADCs)—which comprise a monoclonal antibody linked to a cytotoxic drug via a molecular linker—have demonstrated remarkable efficacy in the treatment of HER2-positive (HER2+) malignant breast carcinoma. These conjugates combine monoclonal antibodies for direct receptor blockade with small molecule cytotoxic drugs to induce apoptosis and inhibit tumor growth 34,35. The mechanisms of action triggered by ADCs include antibody-dependent cellular cytotoxicity (ADCC) 36 and complement-dependent cytotoxicity (CDC), as shown in Figure 3. HER2-targeted ADCs deliver lethal payloads to malignant cells via monoclonal antibodies such as trastuzumab, which binds to HER2, thereby disrupting signaling pathways and hindering tumor progression 37. ADCs utilizing cleavable linkers release their cargo into the tumor microenvironment or within target cells, triggering the bystander effect to eliminate adjacent HER2-negative cells; conversely, non-cleavable or rigid linkers release cargo exclusively within the target cells. Further, ADCs inhibit HER2 shedding and enhance cytotoxicity via ADCC and CDC, thereby increasing therapeutic efficacy 38.
While ADCs are promising therapeutic agents, resistance to trastuzumab-based conjugates remains a major barrier in breast cancer treatment, prompting the development of novel ADCs to mitigate these challenges. In the case of trastuzumab emtansine (T-DM1), resistance occurs via the downregulation of the target antigen (HER2), heterogeneous HER2 expression, or HER2 truncation and heterodimerization, which prevents ADC binding 39. Additionally, even upon successful binding, the receptor-antibody complex may fail to internalize efficiently or may be misrouted within the cell, failing to reach the lysosomes for drug release; such impaired internalization, trafficking, and lysosomal dysfunction result in the loss of cytotoxic efficacy 40. Furthermore, increased expression of ATP-binding cassette (ABC) transporters is linked to T-DM1 resistance, as these transporters efflux excessive drug from the cell 41. Consequently, modifying ADC internalization and trafficking, or inhibiting ABC transporter expression, represent viable strategies to overcome resistance.
DESTINY-Breast01 (NCT03248492), a multicenter, single-arm study funded by Daiichi Sankyo and AstraZeneca, evaluated the safety and efficacy of trastuzumab deruxtecan (DS-8201a) against advanced HER2+ breast cancer 42. The study cohort comprised patients heavily pretreated with a median of six prior therapies, including T-DM1, indicating resistance to existing regimens. The outcomes were significant, with an objective response rate (ORR) of 60.9%. Among these, 6% achieved a complete response, while 54.9% exhibited significant tumor shrinkage, indicating that DS-8201a superior efficacy compared to available therapies. In 2019, it was approved for adults with unresectable or metastatic HER2+ breast carcinoma 42. In DESTINY-Breast03 (NCT03529110), trastuzumab deruxtecan (T-DXd) was compared with T-DM1 in patients previously treated with trastuzumab and a taxane. This Phase III, open-label, randomized trial showed significant improvement in progression-free survival (PFS) with T-DXd; at 12 months, 76% of patients in the T-DXd arm were alive without disease progression, compared to 34.1% in the T-DM1 arm. The overall survival (OS) rate was 94.1% for T-DXd and 85.9% for T-DM1. However, adverse events were more prevalent with T-DXd, including interstitial lung disease (ILD) in 10.5% of patients versus 1.9% for T-DM1. Overall, T-DXd exhibited superior efficacy over T-DM1, representing an effective treatment option for HER2+ breast cancer 43.
The Phase III DESTINY-Breast04 trial was conducted on patients with low HER2 expression levels, for whom most HER2-targeted therapies are typically ineffective. The study examined the efficacy of T-DXd in HER2-low cancers following prior chemotherapy. Results demonstrated improved PFS (10.1 vs. 5.4 months; Hazard Ratio [HR] 0.51; P < 0.001) in the hormone receptor-positive cohort. Similarly, OS was improved (23.9 vs. 17.5 months; HR 0.64; P = 0.003). However, 52.6% of patients experienced serious adverse events, 12% developed ILD, and 0.8% of cases were fatal. Overall, median PFS and OS were improved using T-DXd compared to traditional chemotherapy 43,44. Several other trials, including DESTINY-Breast 05, 06, 08, 09, and 11, are currently ongoing (NCT04622319, NCT0449425, NCT04556773, NCT04784715, NCT05113251).
Similarly, other ADCs such as ARX788 and ZW49 are undergoing Phase I clinical trials. ARX788 is an anti-HER2 ADC conjugated to monomethyl auristatin F (MMAF), a microtubule inhibitor 45, while ZW49 is a bispecific antibody (ZW25) conjugated to monomethyl auristatin E (MMAE), which internalizes rapidly into HER2-expressing cells 46. MEDI4276, another bispecific antibody conjugated to a tubulysin-based microtubule inhibitor, targets HER2-overexpressing cells, including those resistant to T-DM1; however, its Phase I trial was limited by toxicity 47.
SYD985 (trastuzumab duocarmazine) is an ADC combining trastuzumab with a duocarmycin alkylating agent via a cleavable linker. The cleavable linker releases the payload extracellularly, demonstrating strong activity against HER2-low tumors. Phase I trials indicated an ORR of 33% and a median PFS of 9.4 months 48; the Phase III TULIP trial is ongoing. In a late-stage clinical trial comparing T-Duo (SYD985) with physician’s choice (PC) in patients with unresectable advanced carcinomas, 437 patients (291 in the T-Duo arm and 146 in the PC arm) with a median age of 56 were enrolled. The median PFS was 7 months for T-Duo compared to 4.9 months for PC, with a median OS of 20.4 months versus 16.3 months, respectively. The ORR was 27.8% for T-Duo and 29.5% for PC. Although T-Duo improved PFS relative to PC, it exhibited higher ocular toxicity (52.8%) compared to PC (48.2%) 49, highlighting a need for more innovative strategies.
In summary, ADCs for HER2+ breast cancer show variations in efficacy, safety, and resistance governed by molecular design, patient profiles, and clinical settings. Second-generation ADCs like T-DXd have shown superior response rates and PFS compared to T-DM1, as evidenced in the DESTINY-Breast03 trial 43. This superiority is attributed to the higher drug-to-antibody ratio, membrane-permeable payload, and bystander killing effect, notwithstanding the higher risk of ILD. In contrast, T-DM1 maintains activity with mild systemic toxicity. Other agents designed with cleavable linkers (e.g., SYD985) or engineered bispecifics (e.g., ZW49 and MEDI4276) address tumor heterogeneity but are limited by organ-specific toxicities. ABC transporter overexpression, impaired trafficking, and HER2 downregulation contribute to resistance. Overcoming these barriers requires improving internalization and optimizing linkers. Currently, T-DXd is the most effective ADC for both HER2-positive and HER2-low tumors. Future priorities should focus on improving linker-payload stability and mitigating pulmonary toxicity 45,46,47,48.
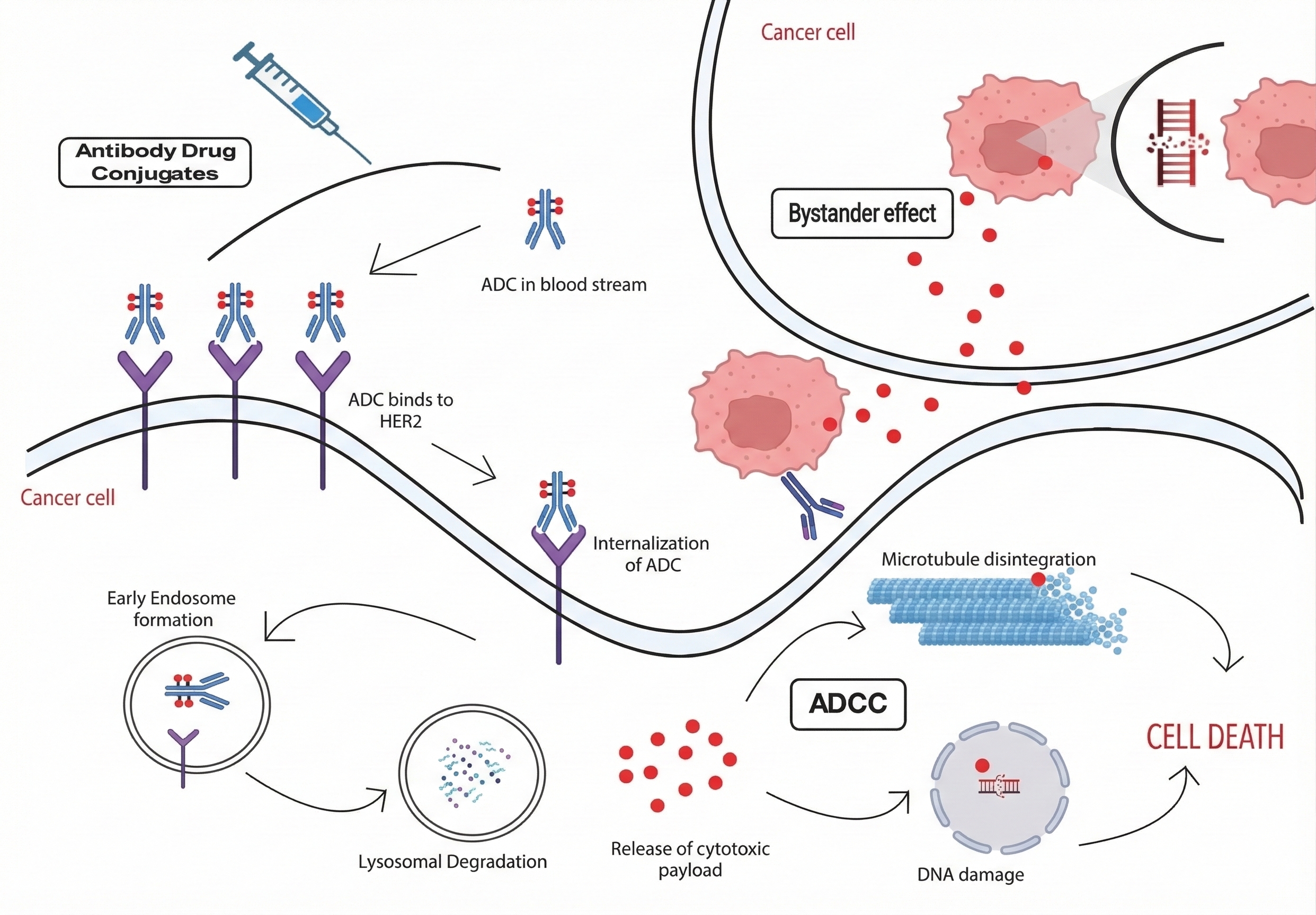
Schematic representation of the mechanism of action and resistance of Antibody-Drug Conjugates (ADCs) in HER2-positive breast cancer. This figure illustrates the targeted delivery of cytotoxic payloads to malignant cells. Upon the binding of the monoclonal antibody (e.g., trastuzumab) to the HER2 receptor, the ADC complex is internalized via receptor-mediated endocytosis, leading to early endosome formation and subsequent lysosomal degradation. The release of the cytotoxic payload (e.g., microtubule inhibitors or DNA-damaging agents) into the cytosol triggers apoptosis. ADCs utilizing cleavable linkers can also exert a bystander effect, where the released payload diffuses to eliminate adjacent HER2-negative cancer cells. Additionally, the figure highlights mechanisms of action such as Antibody-Dependent Cellular Cytotoxicity (ADCC) and Complement-Dependent Cytotoxicity (CDC). Known resistance pathways, including target antigen downregulation, impaired lysosomal trafficking, and ABC transporter-mediated drug efflux, are also contextually represented.
DNA repair: Role of BRCA proteins and PARP enzymes
The nucleus contains a vast array of DNA damage sensors that recruit downstream effectors to sites of injury. One such protein, poly(ADP-ribose) polymerase-1 (PARP1), is ubiquitously expressed and activated by DNA strand breaks. Upon activation, it synthesizes poly(ADP-ribose) (PAR) chains, which serve as a scaffold or signal for the recruitment of numerous DNA repair proteins 34. PARP1 is one of 17 enzymes that transfer PAR or mono-ADP-ribose moieties onto themselves or other target proteins, making it a member of the ADP-ribosyltransferase superfamily. Additionally, PARP1, PARP2, and PARP3 are DNA-dependent ADP-ribosyltransferases. While PARP3 acts primarily as a mono-ADP-ribosyltransferase, PARP1 and PARP2 catalyze the formation of long PAR chains 35.
The covalent attachment of negatively charged PAR to lysine, glutamate, or aspartate residues on target proteins is known as poly(ADP-ribosyl)ation, or PARylation. PARylation can stabilize or destabilize protein-DNA complexes, modulate protein-protein interactions, regulate protein functions, enhance enzymatic activity, and target proteins for proteasomal degradation. Through these mechanisms, PARP proteins regulate various cellular processes, including transcription and DNA replication. They also play a significant role in the DNA damage response (DDR) and cell death pathways 36.
In response to DNA damage, PARP1 can induce a 500-fold increase in cellular PARylation activity, accounting for 85–90% of total cellular PAR. Both PARP1 itself (autoribosylation) and other nuclear acceptor proteins can be modified with 50 to 200 PAR residues. In addition to its role in base excision repair (BER), PARP1 functions as a sensor for single-strand breaks (SSBs) 37. Upon detecting DNA breaks, PARP1 quickly undergoes autoribosylation and facilitates the formation of PAR on both histones and non-histone proteins through a transesterification process, as shown in Figure 4. Furthermore, the recruitment of PARP2 to the damage site is mediated by PARP1-induced PARylation. Subsequently, PARP1 and PARP2 amplify the signal required for the assembly, processing, and eventual repair of DNA damage 38.
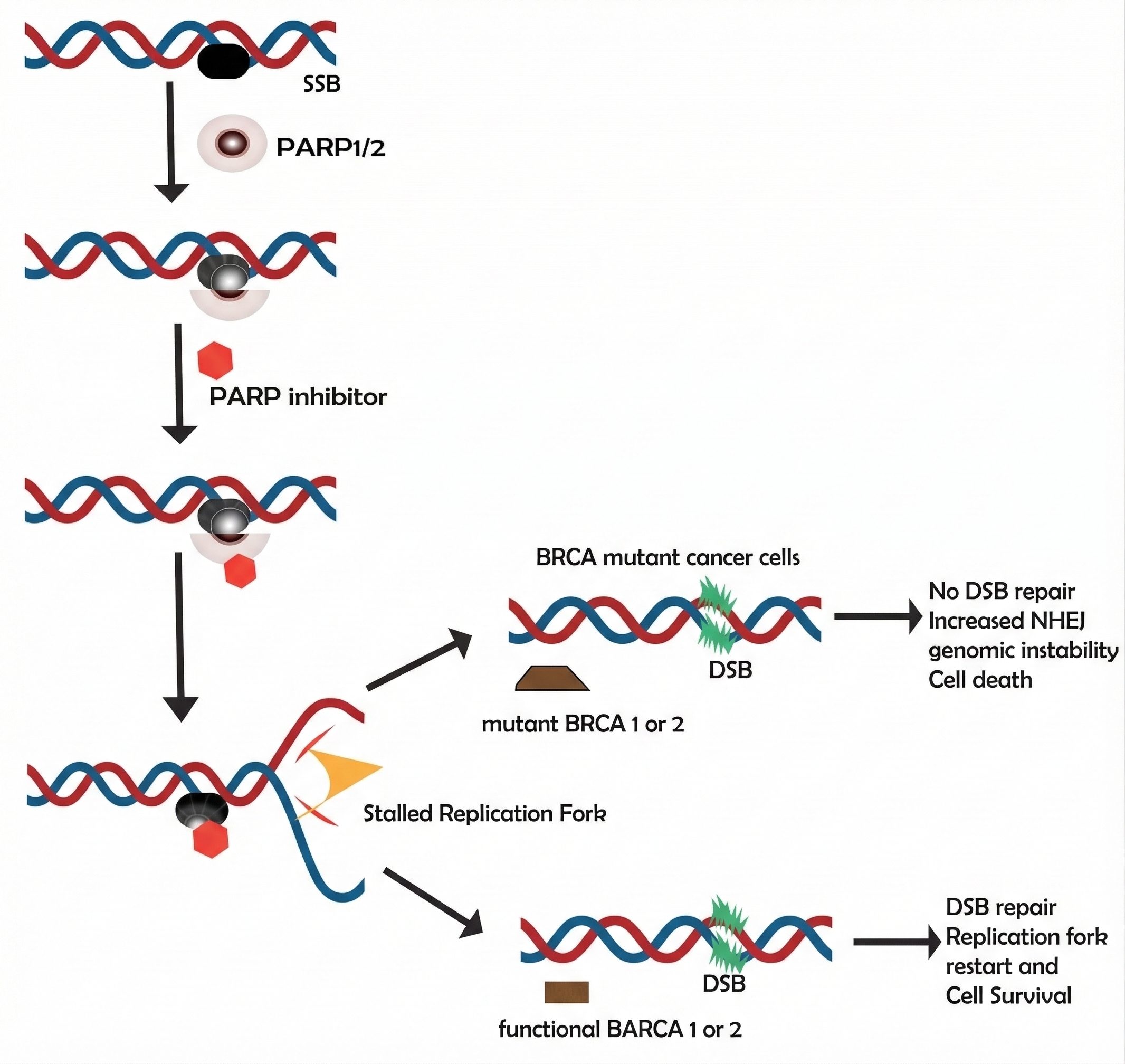
Schematic representation of DNA damage repair and the mechanism of synthetic lethality via PARP inhibition. This figure illustrates the cellular response to DNA damage, where single-strand breaks (SSBs) trigger the recruitment and activation of PARP1/2 enzymes. Under normal conditions, these enzymes facilitate base excision repair (BER). However, in the presence of PARP inhibitors, the enzymes become trapped on the DNA, leading to the stalling of replication forks and the conversion of SSBs into lethal double-strand breaks (DSBs). In BRCA-mutant cancer cells, the absence of functional homologous recombination (HR) prevents the repair of these DSBs, forcing the cell toward genomic instability and apoptosis. Conversely, in cells with functional BRCA1 or 2, the DSBs are successfully repaired through HR, allowing for replication fork restart and cell survival.
Effectiveness of PARP inhibitors in clinical trials
By inhibiting PARP enzyme activity and the subsequent PARylation process, PARP inhibitors (PARPi) are designed to impede DNA repair. Their mechanism of action involves competing with NAD+ for binding to the PARP catalytic domain. In 2005, two independent groups demonstrated that PARP inhibition induces synthetic lethality in BRCA1- and BRCA2-mutant cancers, highlighting a strong synergistic relationship between BRCA function and PARP1 activity 39.
In the presence of PARPi, endogenously produced single-strand breaks (SSBs) are converted into double-strand breaks (DSBs) during cell division due to the impairment of base excision repair (BER). While normal tissues possessing at least one wild-type gene copy can repair DSBs via functional homologous recombination (HR), cells with BRCA dysfunction are unable to repair this damage and undergo apoptosis. In patients with germline BRCA mutations, tumor cells typically exhibit biallelic deficiency and faulty HR, whereas normal tissues maintain BRCA proficiency 40.
Other pathways underlying the anticancer activity of PARPi have recently been identified. A primary mechanism is the "trapping" of PARP1 onto damaged DNA; the resulting PARP-DNA complexes obstruct both transcription and DNA replication. This process is driven by allosteric conformational changes in PARP1 and PARP2 that stabilize their association with DNA, acting independently of catalytic inhibition. Notably, the cytotoxic potency of a PARP inhibitor correlates with its ability to induce PARP1 trapping and the accumulation of these toxic PARP-DNA complexes 41.
Furthermore, BRCA1 mutations that impair heterodimerization with BARD1 lead to aberrant PAR formation and disrupted recruitment of BRCA1/BARD1 complexes to DNA damage sites, ultimately impairing HR-mediated repair and triggering cell death. The PARPi mechanism also involves the activation of non-homologous end joining (NHEJ), because inhibited PARP1 is less effective at preventing KU proteins from interacting with free DNA ends 42. Excessive NHEJ activation leads to genomic instability and eventual cancer cell death. Additionally, inhibition of PARP1 prevents alternative end-joining (Alt-EJ) activation, further promoting apoptosis.
Finally, the effects of PARPi are mediated by the upregulation of death receptors DR5 and FAS via the activation of the SP1 and NF-κB transcription factors. This sensitizes tumor cells to TRAIL or FASL death receptor ligands, as shown in Figure 5. In summary, PARPi induce cell death by trapping PARP1/2 on damaged DNA, inhibiting Alt-EJ-mediated repair in HR-deficient cells, and promoting death receptor upregulation through SP1 and NF-κB signaling 43.
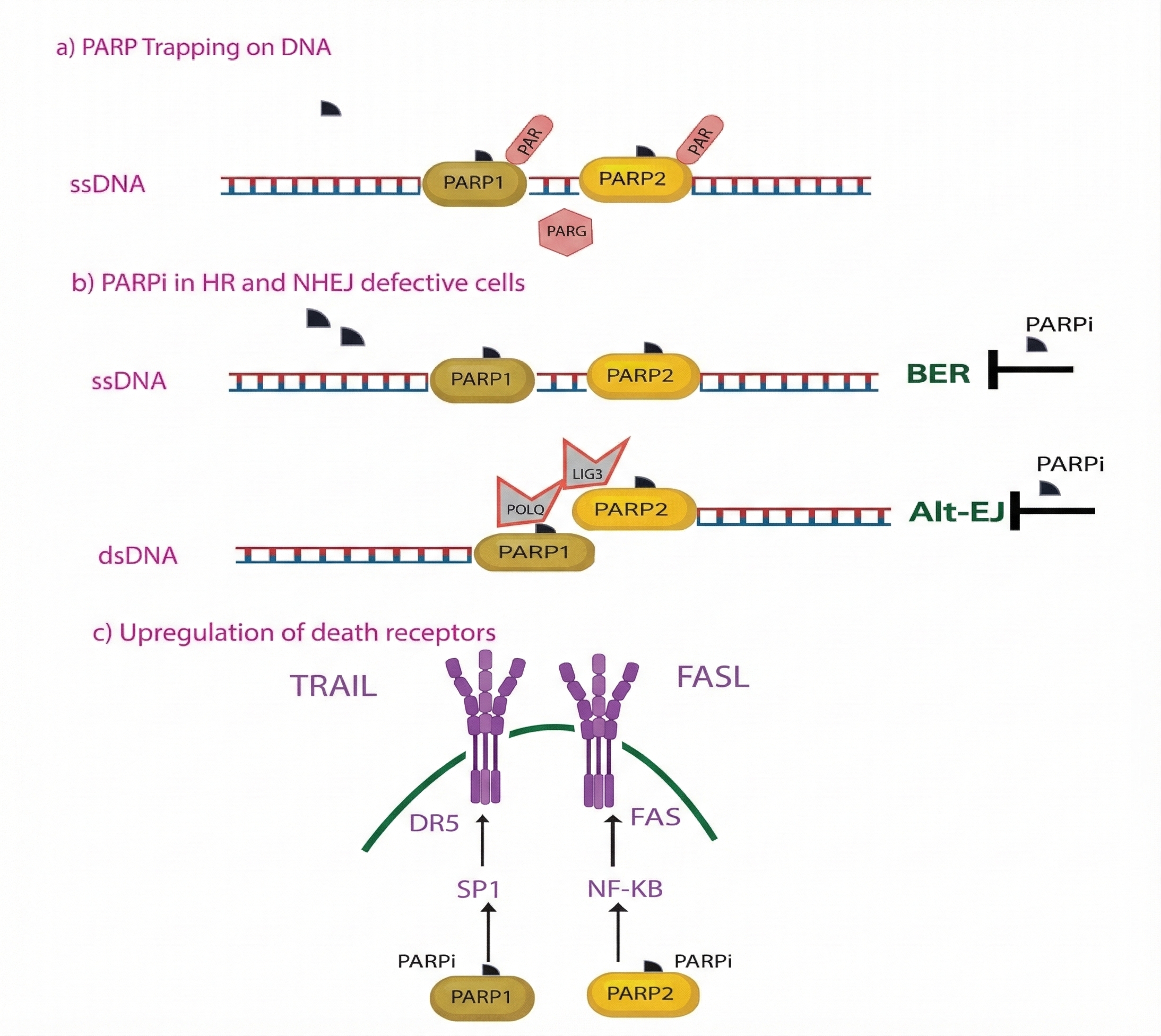
Multi-faceted anticancer mechanisms of PARP inhibitors (PARPi) in DNA-repair-deficient cells. This schematic illustrates three primary pathways through which PARPi induce cytotoxicity: (a) PARP Trapping on DNA: PARPi compete with NAD+ for the catalytic domain, causing an allosteric change that traps PARP1/2 onto damaged DNA, creating toxic complexes that obstruct transcription and replication. (b) Inhibition of Alternative Repair Pathways: In homologous recombination (HR) and non-homologous end joining (NHEJ) defective cells, PARPi block base excision repair (BER) and alternative end-joining (Alt-EJ) mechanisms, preventing the repair of single-strand (ssDNA) and double-strand (dsDNA) breaks. (c) Upregulated Expression of Death Receptors: PARPi treatment triggers the activation of SP1 and NF-κB transcription factors, which upregulate the expression of DR5 and FAS death receptors. This sensitizes tumor cells to extrinsic apoptosis mediated by TRAIL and FASL ligands.
Clinical Developments in breast cancer
In BRCA-mutated cancers, various PARP inhibitors (PARPi) have been studied. Two primary strategies have been employed to expand PARPi clinical applications: PARPi monotherapy in cancers with defective DNA damage repair pathways, and the use of PARPi in combination with DNA-damaging chemotherapy 39. Currently, five PARPi are under investigation in clinical trials: veliparib, olaparib, rucaparib, niraparib, and talazoparib. The agent iniparib was originally developed as a PARPi 40; however, following unfavorable outcomes in a Phase III trial evaluating the drug combined with platinum and gemcitabine for metastatic triple-negative breast cancer (TNBC), subsequent investigations demonstrated that iniparib fails to inhibit PARP in vitro. As tumor sensitivity to platinum agents correlates with the presence of homologous recombination (HR) deficiency, platinum-sensitive tumors are expected to respond to PARPi therapy 41.
Comparative pharmacological potency and clinical trial profiles of various PARP inhibitors (PARPi) in BRCA-positive malignancies. This table delineates the inhibitory factors and cytotoxic mechanisms of five primary PARPi: Niraparib, Veliparib, Talazoparib, Olaparib, and Rucaparib. It categorizes each agent based on its capacity for PARP-DNA complex trapping and its half-maximal inhibitory concentration (IC50) for catalytic inhibition. While Talazoparib is identified as the most potent catalytic inhibitor and "trapper," the table illustrates that a high degree of enzyme inhibition does not always correlate directly with trapping efficiency. The associated clinical trial identifiers (e.g., EMBRACA, OlympiAD, BROCADE3) are provided to link these biochemical properties to their respective therapeutic outcomes.
| Name of the Drug | Inhibitory Factor | PARP-DNA Complexes Trapping | IC50 (PARP Catalytic Inhibition) | Clinical Trial Indentifiers | References |
|---|---|---|---|---|---|
| Niraparib | “PARP 1/2” | ✔ (highest) | ⋍0.57nM | EMBRACA (NCT01945775) | |
| Veliparib | “PARP 1/2” | ✔(strong) | ⋍3.8nM | NOVA (NCT01847274) | |
| Talazoparib | “ARP 1/2” | ✔(strong) | ⋍7.1nM | ARIEL3 (NCT01968213) | |
| Olaparib | “PARP 1/2/3” | ✔(moderate) | ⋍5nM | OlymiAD (NCT02000622) | |
| Rucaparib* | “PARP 1/2/3” | 🞪(weak) | ⋍3.3nM | BROCADE3 (NCT02163694) |
The potency of PARP inhibitors (PARPi) regarding PARP trapping and catalytic inhibition varies significantly, as delineated in Table 3. Notably, the magnitude of PARP enzyme inhibition does not directly correlate with the extent of PARP trapping. Among the clinically available PARPi, talazoparib exhibits the most potent catalytic inhibition, demonstrating approximately 100 times greater efficacy than niraparib. While rucaparib, olaparib, and niraparib similarly induce PARP trapping, talazoparib remains the most potent trapper. Conversely, although veliparib has been reported to induce PARP trapping, this mechanism appears to be least pronounced with this agent 42.
Given that talazoparib exerts the most profound effect, these variations in inhibitory effectiveness and trapping capacity may serve as biomarkers for PARPi cytotoxicity in BRCA-mutant cancers; however, further research is required to determine if these properties translate into superior clinical outcomes. Furthermore, the maximum tolerated dose of monotherapy or combination regimens with chemotherapy may be limited by the increased cytotoxicity associated with PARP trapping, which can exacerbate adverse events, such as cytopenias 49,50.
Clinical trial of PARPi for the treatment of BRCA1/2 mutated cancers
Following the successful development of PARP inhibitors (PARPi) for BRCA-mutated metastatic breast cancer, clinical trials are currently examining their impact in early-stage disease within both neoadjuvant and adjuvant settings. The I-SPY 2 trial is a multicenter, randomized, phase 2 platform utilizing numerous experimental arms to evaluate the addition of new medications or combinations to standard neoadjuvant chemotherapy. The primary endpoint of this trial is pathological complete response (pCR). In one arm, patients with triple-negative breast cancer (TNBC) were randomized to receive either paclitaxel alone or veliparib (50 mg BID for 21 days) in conjunction with carboplatin for four cycles 51.
Following therapy, all patients received four cycles of doxorubicin and cyclophosphamide (AC), followed by surgery. The standard therapy arm (paclitaxel followed by AC) achieved a pCR rate of 26%, whereas the investigational veliparib/carboplatin arm demonstrated an estimated pCR rate of 52%. Pathologic complete response serves as a surrogate endpoint for improved event-free survival in breast cancer. Notably, information regarding germline BRCA mutation status for this cohort has not yet been released.
Subsequently, the BrighTNess Phase 3 trial was designed to evaluate the addition of carboplatin and veliparib to standard neoadjuvant chemotherapy. Researchers randomly assigned stage II-III TNBC patients to receive either carboplatin plus veliparib, carboplatin alone, or placebo 52. Additionally, neoadjuvant talazoparib monotherapy demonstrated significant activity in a pilot phase 2 study. After two months of treatment, 13 patients with BRCA-mutant breast cancer showed a median ultrasound-measured tumor shrinkage of 88% (range 30–98%). This research was terminated early in order to expand the investigation of neoadjuvant talazoparib as a standalone preoperative treatment and further evaluate the pCR rate 53.
The effect of PARP inhibitors on BRCA1 and BRCA2 tumor cells
For patients with BRCA-mutant triple-negative breast cancer (TNBC) or hormone receptor-positive, HER2-negative breast cancer, the phase 3 OlympiA trial evaluated the safety and efficacy of adjuvant olaparib (300 mg BID) versus placebo for a maximum duration of 12 months. Eligible patients were required to have completed neoadjuvant or adjuvant chemotherapy in addition to definitive local therapy. Randomization was stratified by prior neoadjuvant versus adjuvant chemotherapy, previous platinum chemotherapy use, and hormone receptor status 54.
In the post-neoadjuvant cohort, patients without a pathological complete response (pCR) were included. The primary endpoint was invasive disease-free survival, while secondary endpoints included safety and tolerability, overall survival, distant disease-free survival, and the incidence of novel primary cancers. Additionally, olaparib is currently being investigated as a neoadjuvant treatment for basal TNBC and BRCA-mutant breast cancer in combination with platinum-based chemotherapy 55.
Furthermore, a phase 2 trial is evaluating rucaparib as maintenance therapy following cisplatin for TNBC and BRCA-mutant breast cancer with residual disease after neoadjuvant anthracycline- and taxane-based therapy. Following 2 to 4 weeks of cisplatin, patients were randomized to receive cisplatin with or without rucaparib. According to preliminary data, patients treated with an anthracycline-containing regimen showed a significant improvement in two-year disease-free survival with the addition of rucaparib to cisplatin (69.9% vs. 55.9%). Notably, the benefit to disease-free survival was unaffected by BRCA mutation status 56.
Increasing the Clinical Use of PARPi
The concept of synthetic lethality as a therapeutic approach has been validated by the anti-tumor efficacy of PARP inhibitors (PARPis) in BRCA-mutated breast and ovarian carcinomas. Furthermore, evidence of PARPi effectiveness is emerging for other BRCA-mutant malignancies, including prostate and pancreatic cancers. However, it remains to be determined whether this efficacy can be extended to other tumor types lacking germline BRCA1/2 mutations that harbor other defects in DNA damage repair pathways 57.
Recent research has increasingly focused on cancers displaying the "BRCAness" phenotype. "BRCAness" refers to malignant tumors that do not harbor germline BRCA1 or BRCA2 mutations but share clinical and molecular characteristics of homologous recombination (HR) repair failure due to distinct genetic alterations. These include promoter hypermethylation of BRCA1, somatic mutations in BRCA1 or BRCA2, or alterations in other genes involved in double-strand break (DSB) repair. Mirroring BRCA-associated tumors, these cancers exhibit sensitivity to platinum-based chemotherapy 58. Currently, a standardized biomarker for "BRCAness" does not exist, though extensive research has aimed to develop classifiers to predict susceptibility to PARP inhibition. Within the context of the I-SPY2 clinical trial, Severson et al. recently developed a 77-gene signature to define "BRCAness" and evaluate its utility in predicting responses to neoadjuvant veliparib combined with carboplatin 34.
Despite the small sample size, they demonstrated a correlation between the "BRCAness" classification and patient response within the veliparib/carboplatin arm. To quantify "BRCAness," studies are also utilizing an HR deficiency (HRD) score that integrates three distinct indicators of genomic instability: large-scale state transitions, telomeric allelic imbalance, and loss of heterozygosity. Patients with a favorable score (≥42) exhibited improved responses to platinum therapy. This hypothesis is currently being investigated further using other PARPis, such as rucaparib and talazoparib 37. Additionally, clinical investigations are exploring PARPi combinations with immunotherapies, including CTLA-4 and PD-1/PD-L1 antibodies. The enhanced genomic instability in HRD tumors may be linked to an increased neoantigen load, which is associated with a more robust immune response. Supporting this combination strategy, preliminary evidence indicates that PARPis may sensitize tumors to immunotherapy 36.
Moreover, preliminary data suggest that DNA-repair-defective tumors selectively activate the cell cycle-specific STING (stimulator of interferon genes)-dependent innate immune response. Early-phase I studies are assessing the efficacy of the anti-angiogenic agent cediranib or the PD-L1 inhibitor durvalumab in combination with olaparib in women with progressive solid tumors. In a cohort of twelve evaluable patients receiving durvalumab plus olaparib, eight experienced stable disease for more than four months and two achieved a partial response, resulting in an 83% disease control rate. Consequently, trials investigating the combination of PARPis and PD-L1 inhibitors are currently underway 59.
PI3K inhibition represents another strategy to expand the utility of PARP inhibition beyond BRCA-mutant breast tumors. The PI3K/MAPK pathway facilitates DNA break repair within tumor cells in addition to its pro-proliferative and anti-apoptotic roles. In preclinical breast cancer models, the combination of olaparib and a PI3K inhibitor slowed tumor growth, indicating synergy in BRCA-mutant cancers. Preliminary clinical data demonstrated success with the combination of olaparib and the PI3K inhibitor buparlisib (BKM120) in patients with triple-negative breast cancer (TNBC) or ovarian cancer, yielding an overall response rate (ORR) of 24% (10 of 42 patients) 60.
PARPi Resistance
Malignancies may develop resistance to poly (ADP-ribose) polymerase inhibitor (PARPi) therapy, analogous to the development of resistance to other targeted treatments. The restoration of the homologous recombination (HR) repair pathway is one recognized strategy underlying this phenomenon. Reversion mutations that restore BRCA1/2 function and confer PARP resistance have been shown to arise from the enhanced genomic instability associated with PARP inhibition 37,61.
Furthermore, the loss of certain DNA repair proteins, such as REV7 and p53-binding protein 1 (53BP1), can partially restore HR activity in BRCA-deficient cells. Another mechanism of resistance is increased drug efflux mediated by the upregulation of P-glycoprotein expression, which lowers intracellular PARPi concentrations. Additionally, resistance to PARPi can result from variations in PARP1 expression levels within cancer cells.
Elevated PARP1 expression is linked to reduced survival in patients with breast cancer and contributes to PARPi resistance. Although the extent of overlap remains unclear, PARPi and platinum chemotherapy share several potential resistance mechanisms. In platinum-resistant scenarios, these mechanisms may restrict the clinical utility of PARPi; consequently, alternative strategies to counteract acquired resistance are required. Notably, most current PARPi studies exclude individuals with platinum-resistant disease 38.
PARPi for breast cancer approved recently
A Phase I clinical trial demonstrated that a synthetic lethality approach could be successfully implemented by showing that the PARP inhibitor (PARPi) olaparib, which inhibits PARP1 and PARP2, exhibited objective antitumor activity in patients with BRCA-mutated breast, prostate, or ovarian cancers, while demonstrating a more favorable side-effect profile than conventional chemotherapies. Subsequent trials verified that PARPis are more effective as monotherapy for BRCA-mutated ovarian cancer than for BRCA wild-type tumors 59.
In 2014, the European Commission approved olaparib (capsule) for patients with recurrent, high-grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancers who achieved a complete or partial response following platinum-based chemotherapy. The FDA also approved olaparib to treat germline BRCA-mutated advanced ovarian cancer (OC) in patients who had received three or more prior lines of chemotherapy. Subsequently, both the FDA and EMA approved a tablet formulation of olaparib, which reduces the daily pill burden, for maintenance treatment of platinum-sensitive recurrent OC, irrespective of BRCA mutation status 34.
According to Robson and Tung 60, the OlympiAD trial led to the approval of olaparib for patients with previously treated, HER2-negative metastatic breast cancer. Researchers analyzed 302 patients with hereditary BRCA mutations and HER2-negative status, including those with triple-negative disease. Each participant received either olaparib or single-agent chemotherapy (capecitabine, eribulin, or vinorelbine). After a median follow-up of 14.5 months, olaparib exhibited a significantly higher progression-free survival (PFS) rate than conventional treatment 42.
Although PFS improved by only several months, the clinical outcomes remain optimistic. However, the OlympiAD study was unable to evaluate the relative efficacy of PARPis compared to platinum-based chemotherapy in patients with germline BRCA mutations, as PARPi efficacy was not directly compared to platinum compounds. Therefore, additional research is needed to compare PARPis with platinum-based agents in germline BRCA-mutated breast cancer. Furthermore, it remains unknown whether PARPi treatment is efficacious in HER2-negative breast cancer harboring somatic BRCA mutations 61.
Among PARP inhibitors, talazoparib possesses the highest cytotoxicity and the strongest PARP-trapping ability. In contrast, olaparib and niraparib exhibit better tolerability and moderate potency. Rucaparib is effective in combination regimens but demonstrates less catalytic inhibition. PFS outcomes in clinical trials (such as OlympiAD, EMBRACA, and BROCADE3) are superior to those of chemotherapy. Due to variations in patient selection and study design, survival benefits vary among PARP inhibitors.
Talazoparib is associated with higher hematologic toxicity, including neutropenia and anemia, whereas niraparib and olaparib are better tolerated for long-term therapy. Resistance is primarily driven by BRCA reversion mutations that restore DNA repair capacity. Resistance is also mediated by alterations in PARP1 expression and the overexpression of P-glycoprotein. Notably, many platinum resistance mechanisms overlap with PARPi resistance. Variability in clinical outcomes is explained by patient cohorts, BRCA mutation types, and previous platinum exposure.
The antitumor response is improved when PARPis are combined with PI3K or immune checkpoint inhibitors. In addition to delaying the onset of resistance, combined regimens enhance synthetic lethality. While BRCA-mutant tumours still require PARPi monotherapy, the goal of ongoing research is to extend these advantages to "BRCAness" and other homologous recombination (HR)-deficient subtypes.
Emerging RNA and Epi-transcriptomic Targets:
Recent work by Liu and Lyu 62 identifying ALKBH5 as a driver of resistance to HER2-directed therapy provides compelling proof-of-concept that RNA epitranscriptomic regulators can reprogram tumor metabolism and alter drug response. In preclinical models, ALKBH5 upregulation in HER2-targeted therapy-resistant cells promotes the mA demethylation of GLUT4 mRNA, thereby increasing its transcript stability and protein expression. This phenotype shifts tumor cells toward glycolytic metabolism and reduces susceptibility to anti-HER2 agents. While Liu et al. (2022) demonstrated this ALKBH5–GLUT4–glycolysis axis in vitro and in vivo and reported clinical correlations with poor response, independent validation across additional patient-derived xenografts (PDXs), organoid models, and retrospective clinical cohorts is required to establish its prevalence and prognostic value.
Beyond ALKBH5, the broader family of mA regulators—comprising writers (the METTL3/METTL14 complex), erasers (ALKBH5, FTO), and readers (YTH-domain proteins, IGF2BPs)—modulates key cancer hallmarks, including proliferation, alternative splicing, metabolism, immune evasion, and therapy sensitivity 63. Mechanistic studies and recent reviews demonstrate that dysregulated mA deposition and removal can reprogram transcripts governing DNA repair, metabolic enzymes, and pro-survival factors; this reprogramming frequently correlates with resistance to chemotherapy and molecularly targeted agents. Integrating METTL3 and FTO transitions this section from a single-study observation to a substantiated, clinically relevant theme regarding epitranscriptomic regulation in breast cancer 64.
METTL3, the principal catalytic “writer” in the METTL3–METTL14 complex, exerts oncogenic functions by promoting $m^6A$ methylation on transcripts that regulate cell growth, alternative splicing, and therapy response. METTL3-dependent $m^6A$ marks can increase the translation or stability of oncogenic mRNAs and influence splicing decisions that favor malignant phenotypes. There is emerging evidence that METTL3 activity supports homologous recombination (HR) repair programs and specific stem-like tumor states, which have direct implications for PARP inhibitor sensitivity 65. The discovery and characterization of STM2457, a selective catalytic METTL3 inhibitor, provides proof-of-concept that pharmacologic inhibition of writers is feasible and therapeutically active in vivo, although data in solid tumors—including breast cancer—remain in the early stages and require focused preclinical and clinical translation 66.
FTO, an alternative mA demethylase, similarly warrants discussion; FTO overexpression has been associated with enhanced proliferation, stemness, and resistance phenotypes. Structure-guided inhibitors, such as FB23 and FB23-2, have demonstrated on-target activity in preclinical models and can phenocopy FTO depletion, thereby reducing proliferation and re-sensitizing cells to cytotoxic agents 67. Importantly, ALKBH5 and FTO can exhibit overlapping, context-dependent, and sometimes opposing effects depending on substrate specificity and tumor lineage 68. Consequently, a comparative analysis of their expression, substrates, and functional impact is essential to clarify which patients might benefit from demethylase-targeted strategies.
Functional roles and therapeutic targeting of key m6A RNA-modifying enzymes in breast cancer. This table summarizes the biological impact of the m6A "writers" (e.g., METTL3), "erasers" (e.g., ALKBH5, FTO), and "readers" (e.g., YTHDF1) on tumor progression and therapy resistance. It highlights specific oncogenic mechanisms, such as the ALKBH5–GLUT4–glycolysis axis which promotes resistance to HER2-targeted agents, and the role of METTL3 in supporting homologous recombination (HR) repair. Additionally, the table lists available pharmacologic probes and first-in-class inhibitors, such as STM2457 and FB23-2, currently utilized in preclinical models to validate these regulators as viable precision medicine targets.
| Regulator | Role | Key reported effects in breast cancer | Therapeutic probes / inhibitors (preclinical / clinical) | References |
|---|---|---|---|---|
| METTL3 | Writer (m⁶A methyltransferase) | Promotes oncogenic mRNA translation; implicated in homologous recombination and therapy resistance; supports stem-like states | STM2457 (first-in-class METTL3 inhibitor, preclinical) | |
| ALKBH5 | Eraser (m⁶A demethylase) | Upregulated in HER2-resistant models; stabilizes GLUT4 mRNA → glycolysis → ADC/TKI resistance | Genetic knockdown/CRISPR (preclinical); no selective clinical inhibitors yet | |
| FTO | Eraser (m⁶A demethylase) | Associated with proliferation, stemness and resistance phenotypes; modulates oncogenic transcript stability | FB23, FB23-2, MA2 (preclinical probes) | |
| YTHDF1 / IGF2BP1 | Readers | Promote translation/stability of oncogenic transcripts; implicated in stress responses and therapy tolerance | No clinical agents yet; research-stage modulation |
Recent Landmark Trials (2022–2024): Practice-Shifting Evidence and Implications
By redefining HER2 categorization beyond immunohistochemistry (IHC) 3+, the DESTINY-Breast04 (2022) trial established trastuzumab deruxtecan (T-DXd) as the first effective therapeutic in HER2-low breast cancer. In hormone receptor-positive (HR+), HER2-low patients, the trial demonstrated significantly improved overall survival (23.9 vs. 17.5 months) and progression-free survival (10.1 vs. 5.4 months) compared with physician's choice chemotherapy. By establishing a new HER2-low therapeutic paradigm and extending eligibility to patients previously classified as HER2-negative, this study transformed clinical practice. In the adjuvant setting for high-risk HER2-positive early breast cancer 70, the ongoing DESTINY-Breast05 trial (2024) directly compares T-DXd with trastuzumab emtansine (T-DM1) to determine whether T-DXd should replace T-DM1 as the standard post-neoadjuvant therapy. A shift in the adjuvant standard of care may be warranted if mature results validate safety, as early data suggest improved invasive disease-free survival and decreased recurrence with T-DXd. Collectively, these DESTINY trials represent a translational milestone, as they validate T-DXd across the spectrum of HER2 expression and support its utility from metastatic to early-stage disease 71.
In HR+/HER2- metastatic breast cancer following resistance to aromatase inhibitors, the CAPItello-291 (2023) trial demonstrated that fulvestrant combined with capivasertib, a potent AKT inhibitor, significantly improved outcomes. The median progression-free survival doubled compared with placebo-fulvestrant (7.2 vs. 3.6 months; HR 0.60, p < 0.001), with clinical benefit observed in both the wild-type and PI3K/AKT/PTEN-altered cohorts 72. These findings extend the landscape of precision medicine beyond PI3K inhibitors (e.g., alpelisib) and validate AKT pathway inhibition as a clinically relevant therapeutic target. Furthermore, the trial supports molecular testing for AKT-pathway alterations to enhance patient selection and refine combination strategies with endocrine therapy. The results of CAPItello-291 have influenced clinical guidelines, establishing capivasertib plus fulvestrant as a new standard of care for HR+/HER2- disease progressing after endocrine therapy 73.
In BRCA-mutated or homologous recombination (HR)-deficient breast cancer, the KEYLYNK-BRCA trials (2023–2024) investigated combined PARP and immune checkpoint inhibition, specifically olaparib plus pembrolizumab or durvalumab. Compared with PARP inhibitor monotherapy, preliminary results indicate extended PFS and sustained responses, especially in the "BRCAness" and PD-L1-positive populations. Combination therapy potentially overcomes resistance associated with PARPi-induced restoration of homologous recombination by enhancing immune activation and DNA-damage signaling 74. These findings suggest a synergistic paradigm integrating immune modulation and DNA repair inhibition to mitigate PARPi resistance. Ongoing Phase III expansions will validate the survival benefit in BRCA-mutant and HR-deficient breast cancer and may reshape maintenance and metastatic treatment algorithms 75.
Collectively, the KEYLYNK-BRCA, CAPItello-291, and DESTINY-Breast04/05 trials represent significant oncological advances from 2022 to 2024, shifting the paradigm of precision oncology from receptor-driven approaches toward molecularly integrated treatment strategies. These studies demonstrate how the integration of immune modulation, targeted pathway inhibition, and genomic profiling expands therapeutic reach, enhances treatment durability, and mitigates resistance. For completeness, these data should be incorporated into tables summarizing pivotal trials, alongside key outcomes (progression-free survival [PFS], overall survival [OS], and objective response rate [ORR]), National Clinical Trial (NCT) identifiers, and publication years. The influence of these seminal studies on redefining patient stratification, treatment sequencing, and future combination strategies in breast cancer management should be explicitly addressed in the discussion and conclusion sections.
Conclusion
Breast cancer treatment has evolved significantly, notably through the incorporation of genomic profiling and genetic analyses, which have enhanced personalized therapeutic strategies. Genomic profiling identifies specific mutations and aberrations in breast cancer, enabling highly tailored treatment modalities. In advanced breast carcinomas, research indicates that the human epidermal growth factor receptor 2 (HER2), ALKBH5 enzyme, BRCA proteins, and poly (ADP-ribose) polymerase (PARP) enzymes are frequently overexpressed. Specifically, HER2-targeted therapies and PARP inhibitors (PARPi) effectively target disease progression in molecularly defined subtypes. HER2-targeted therapies, including monoclonal antibodies and tyrosine kinase inhibitors (TKIs), have substantially improved overall survival for patients with HER2-positive breast carcinoma by increasing pathological complete response rates and lowering recurrence risks.
HER2 (ERBB2) is highly expressed in approximately 15-20% of breast tumors and is linked to aggressive biological behavior. HER2-targeted agents—such as the monoclonal antibodies trastuzumab (which binds to the extracellular HER2 receptor to block downstream signaling) and pertuzumab (which acts synergistically with trastuzumab by inhibiting receptor dimerization)—significantly improve outcomes. Furthermore, TKIs such as lapatinib and neratinib block the intracellular tyrosine kinase domain of HER receptors, thus inhibiting tumor cell proliferation. Similarly, antibody-drug conjugates (ADC)s employ a sophisticated mechanism to target HER2-positive cells; they combine the specificity of monoclonal antibodies with potent cytotoxic payloads. However, resistance to these conjugates (trastuzumab derivatives) remains a major barrier, prompting the development of next-generation ADCs to circumvent these challenges.
Mutations in either the BRCA1 or BRCA2 tumor suppressor genes predispose individuals to malignancy. These mutations cause a significant increase in risk due to impairment of error-free DNA repair processes (homologous recombination). Precision oncology—a rapidly evolving paradigm—takes into account each tumor's unique cellular and molecular characteristics. The foundation of this approach is the ability to accurately stratify patient subpopulations most likely to benefit from tailored interventions and to characterize actionable genomic alterations. Inhibitors of PARP, a family of proteins integral to the machinery involved in the DNA damage response (DDR), are among the most promising therapeutic agents.
While the targeting of estrogen receptors and HER2 has led to numerous advances, treatment options for triple-negative breast cancer (TNBC) remain limited. Designing clinical trials for TNBC patients is challenging because of the disease's marked heterogeneity. To identify subsets of TNBC tumors responsive to chemotherapy or to integrate targeted biological agents, validated predictive biomarkers and accurate tumor categorization are essential. Several PARP inhibitors are currently approved for the treatment of recurrent ovarian cancer harboring BRCA mutations and/or HER2-negative metastatic breast cancer associated with germline BRCA mutations. BRCA-mutant tumors are more likely to respond to PARPi treatment than tumors containing wild-type BRCA due to the principle of synthetic lethality.
Identifying new loci for breast cancer susceptibility will elucidate mechanisms of oncogenesis and provide new therapeutic targets. To achieve true personalized medicine, advances across multiple disciplines are required. This includes the identification of novel susceptibility loci, understanding the role of epigenetics in carcinogenesis, and the discovery of effective therapeutic targets with reduced systemic toxicity. Furthermore, developing prognostic and predictive biomarkers will necessitate transdisciplinary systems biology approaches. As clinical trials incorporate next-generation sequencing (NGS), early findings suggest promising clinical benefit in subsets of advanced cancer patients. In the coming years, it is imperative to implement clinical trials incorporating genomic sequencing and establish shared databases to consolidate genomic information.
Despite the progress in genomic profiling and targeted therapies, several critical gaps remain. First, the mechanisms underlying antibody-drug conjugate (ADC) resistance are still poorly understood. Resistance arises through multiple pathways, including the downregulation or heterogeneous expression of HER2, impaired internalization and lysosomal trafficking, increased drug efflux via ABC transporters, and the activation of bypass signaling pathways such as PI3K/AKT/mTOR. Deeper translational studies using patient-derived xenografts (PDX) and single-cell multi-omics are urgently needed to dissect these mechanisms. Second, HER2 heterogeneity poses a major therapeutic challenge: tumors with variable HER2 expression often respond inconsistently to targeted agents, yet standardized tools to quantify and monitor this heterogeneity are lacking. Third, TNBC remains a therapeutic frontier due to the paucity of robust biomarkers. Future studies should prioritize identifying predictive biomarkers (e.g., DNA repair deficiency signatures and immune-related markers) to stratify TNBC patients. Fourth, preclinical models often fail to recapitulate the human tumor microenvironment (TME), leading to a translational gap. Development of advanced models such as 3D organoids and humanized mouse models could bridge this gap. Fifth, toxicity prediction remains inadequate, as several next-generation ADCs exhibited unexpected adverse effects in early clinical trials. Integrating AI-driven toxicity modeling and pharmacogenomics could improve prediction accuracy. Future directions include site-specific ADC engineering, novel immune-stimulatory conjugates, and rational drug combinations (e.g., PARP inhibitors combined with immune checkpoint blockade) to overcome resistance. Moreover, multi-omics integration with AI-powered analytics and adaptive clinical trial frameworks will be indispensable to accelerating precision oncology in breast cancer.
Abbreviations
ABC: ATP-binding cassette; AC: Doxorubicin and cyclophosphamide; ADC: Antibody-drug conjugate; ADCC: Antibody-dependent cellular cytotoxicity; Alt-EJ: Alternative end-joining; BER: Base excision repair; CDC: Complement-dependent cytotoxicity; CNS: Central Nervous System; CTC: Circulating tumor cell; ctDNA: Circulating tumor DNA; DDR: DNA damage response; DSB: Double-strand break; ECD: Extracellular domain; EGFR: Epidermal growth factor receptor; EMA: European Medicines Agency; FDA: Food and Drug Administration; GLUT4: Glucose transporter type 4; HER2: Human Epidermal Growth Factor Receptor 2; HR: Hormone receptor; HR: Homologous recombination; HRD: Homologous recombination deficiency; IHC: Immunohistochemistry; ILD: Interstitial lung disease; mAb: Monoclonal antibody; MBC: Metastatic breast cancer; MTD: Maximum tolerated dose; NGS: Next-generation sequencing; NHEJ: Non-homologous end joining; ORR: objective response rate; OS: Overall survival; PAR: Poly(ADP-ribose); PARP: Poly(ADP-ribose) polymerase; PARPi: PARP inhibitor; pCR: Pathological complete response; PFS: Progression-free survival; PRISMA: Preferred Reporting Items for Systematic Reviews and Meta-Analyses; SSB: Single-strand break; T-DM1: Ado-trastuzumab emtansine; T-DXd: Trastuzumab deruxtecan; TKI: Tyrosine kinase inhibitor; TME: Tumor microenvironment; TNBC: Triple-negative breast cancer; WHO: World Health Organization.
Acknowledgments
None.
Author’s contributions
All authors equally contributed to this work, read and approved the final manuscript.
Funding
None.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have not used generative AI (a type of artificial intelligence technology that can produce various types of content including text, imagery, audio and synthetic data.
Competing interests
The authors declare that they have no competing interests.