

Evaluating IFN-γ ELISpot in a pediatric case: Unraveling X-linked leaky severe combined immunodeficiency and Omenn syndrome
- Primary Immunodeficiency Diseases Group, Department of Clinical Medicine, Advanced Medical and Dental Institute/Pusat Pakar Klinikal dan Penyelidikan Kanser (PPKPK), Universiti Sains Malaysia, Bertam 13200 Kepala Batas, Pulau Pinang, Malaysia
- Department of Paediatrics, Universiti Kebangsaan Malaysia Medical Center, Hospital Pakar Kanak-Kanak UKM, 56000 Kuala Lumpur, Malaysia
Abstract
Background: Mutations in the interleukin-2 receptor γ-chain gene (IL2RG) cause the X-linked T–B+ severe combined immunodeficiency (X-SCID), also termed common γ (γc) chain deficiency. Hypomorphic IL2RG variants that diminish surface expression of the γc molecule (CD132) can produce atypical clinical and immunological phenotypes. Affected patients typically display absent or markedly reduced T- and natural-killer (NK)-cell numbers while retaining normal B-cell counts.
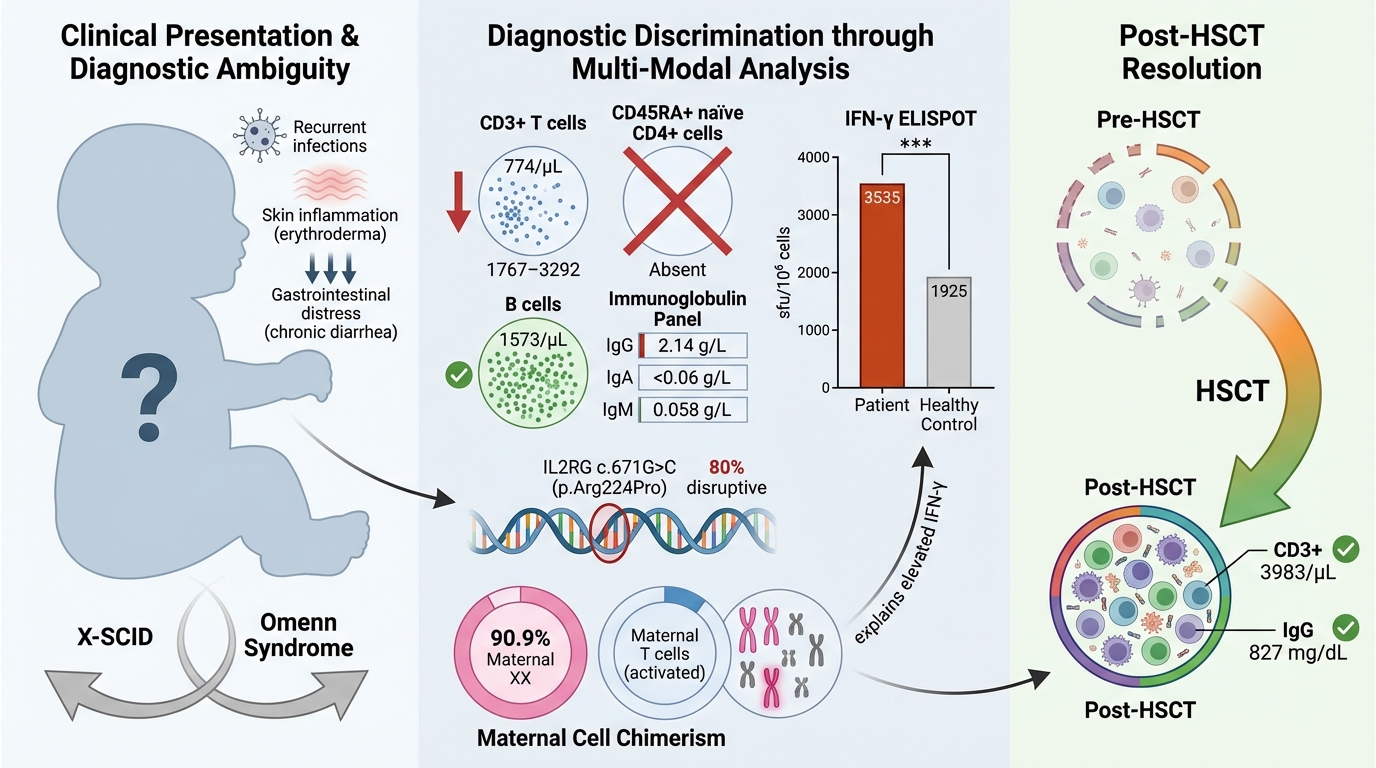
Case presentation: We describe a three-month-old boy who developed recurrent bacterial and viral infections, erythroderma, and chronic diarrhoea. The initial differential diagnosis comprised leaky SCID with Omenn syndrome and SCID with presumed transplacentally acquired maternal T-cell engraftment (TME) and graft-versus-host disease (GVHD). Comprehensive immunological work-up and targeted genetic analysis confirmed X-SCID.
Conclusion: This report underscores the importance of prompt recognition and systematic evaluation of infants with recurrent infections, erythroderma, and chronic diarrhoea. Overlapping features of Omenn syndrome and maternal T-cell engraftment may obscure the diagnosis, highlighting the need for early immunological and molecular investigations. Timely identification of X-SCID is crucial to improve survival and prevent irreversible sequelae. The case supports universal newborn SCID screening and a multidisciplinary approach to the management of complex primary immunodeficiencies.
INTRODUCTION
Patients with X-linked severe combined immunodeficiency (X-SCID) produce markedly reduced numbers of T cells and natural killer (NK) cells. The disease is caused by pathogenic variants in the gene encoding the common γ chain (γc) of the interleukin-2 receptor, which is essential for thymic T-cell development and for the homeostasis and suppressive activity of regulatory T cells. Consequently, affected infants are extremely susceptible to recurrent, severe infections. Because it is X-linked, nearly all patients are male 1.
Classically, γc-deficient patients exhibit normal B-cell numbers but an absence of T and NK cells. Variant immunophenotypes may occur depending on the specific mutation; nonetheless, the T–B+NK– profile remains the most frequent. Clinically, affected infants usually present within the first months of life with failure to thrive, chronic diarrhoea, and recurrent viral, bacterial, and fungal infections 2. Without early intervention—most commonly hematopoietic stem-cell transplantation (HSCT) combined with intravenous immunoglobulin (IVIG)—children with X-SCID rarely survive beyond the first year of life 3. Timely immunological evaluation and molecular confirmation are therefore critical to optimise survival.
We describe a male infant initially suspected of having leaky SCID with Omenn syndrome, or SCID complicated by transplacental maternal engraftment (TME) and graft-versus-host disease (GVHD), owing to his presentation with erythroderma, chronic diarrhoea, and multiple viral and bacterial infections. Immunological evaluation supported this suspicion, and IVIG replacement, antimicrobial prophylaxis, and strict isolation were instituted pending genetic results.
Genetic analysis confirmed X-SCID, and functional assessment with interferon-γ enzyme-linked immunospot (IFN-γ ELISPOT) assays demonstrated residual T-cell activity. These findings underscore the value of early inborn error of immunity (IEI) screening, prompt supportive therapy, and comprehensive immunological work-up in infants with recurrent infections, chronic diarrhoea, and growth failure.
CASE PRESENTATION
The patient was a 3-month-old Indonesian boy born to non-consanguineous parents. He was delivered at full term as the third child and first-born son in a family of three siblings. He had a history of frequent watery stools since birth, which were non-mucoid and yellow in color. At 3 weeks of age, he developed progressive, generalized erythroderma accompanied by desquamation. By 2 months of age, he developed persistent purulent otorrhea from both ears, with minimal improvement despite a course of oral second-generation cephalosporins. He also exhibited fever, rhinorrhea, stertorous breathing, and cough. Despite these symptoms, he was thriving, weighing 6.4 kg (75th percentile) (Figure 1) 4, while receiving mixed breast- and formula-feeding.
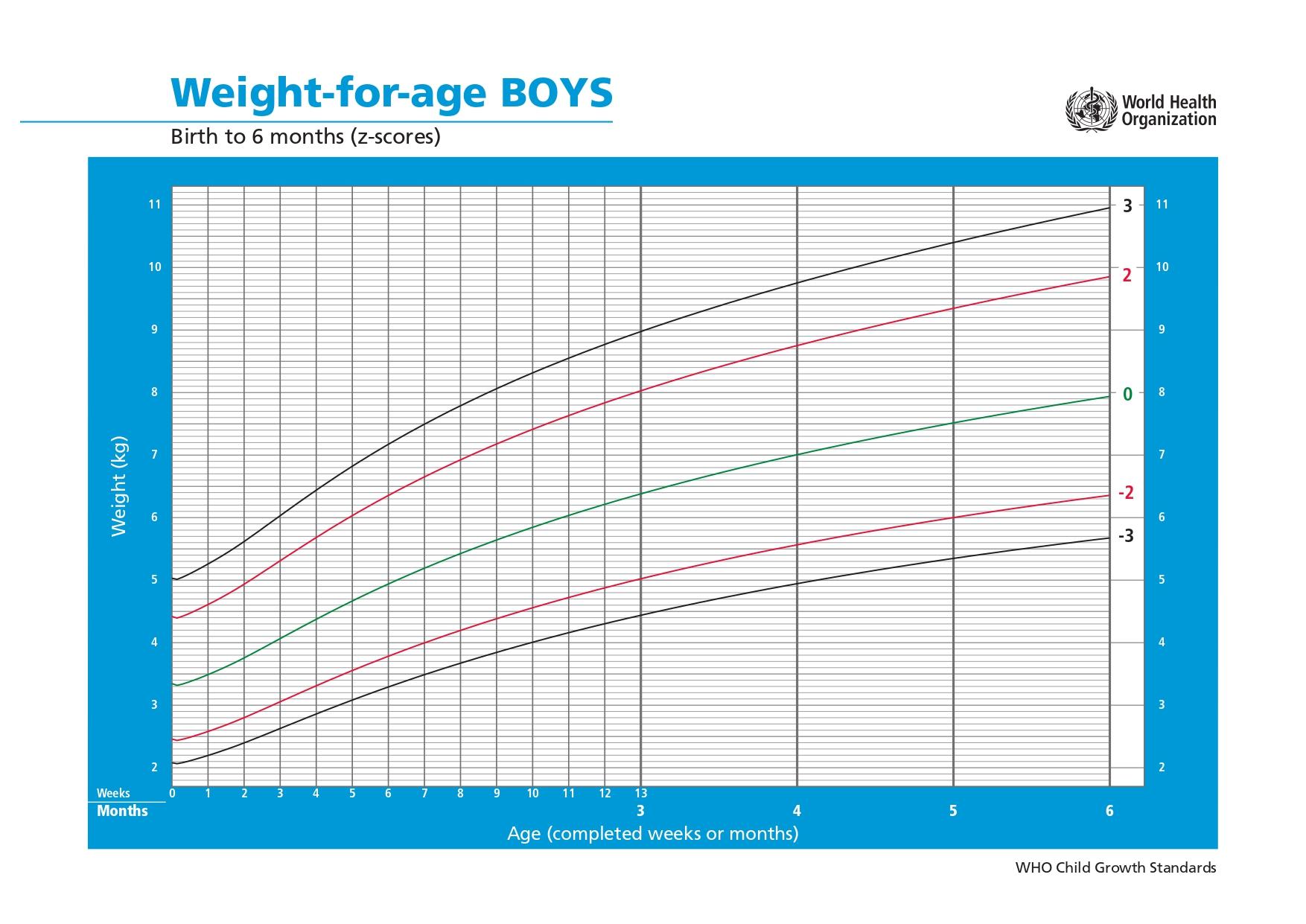
Weight-for-age growth trajectory of the patient based on the World Health Organization (WHO) Child Growth Standards chart for boys from birth to 6 months (weight-for-age z-scores) (4). At age 3months, the patient’s recorded weight of 6.4 kg corresponds to approximately the 75th percentile (between the 0 and +2 z-score curves).
On presentation at 3 months of age, he demonstrated generalized xerosis and erythroderma with palmoplantar desquamation. He also had thick, adherent scales over the scalp and periauricular regions. He exhibited significant nasal congestion, while pulmonary examination remained normal. Otoscopy demonstrated cloudy, yellow discharge in both external auditory canals. There was no oral thrush or ulcers, and no detectable lymphadenopathy or hepatosplenomegaly.
Immunological investigations revealed significant T- and NK-cell lymphopenia, with an absence of naïve CD4⁺ cells. Serological analysis demonstrated pan-hypogammaglobulinemia despite a normal B-cell count (Table 1). A nasal swab culture was positive for rhinovirus/enterovirus, whereas culture of ear discharge yielded a mixed bacterial flora, including Pseudomonas aeruginosa. Urine culture yielded significant growth of Enterococcus faecalis (Table 2).
List of immunological test results
| Immunological tests | Results | Reference Range |
|---|---|---|
| FBC |
Hb 9.7g/dL, Neutrophils 4.05 x 109/L, Lymphocytes 2.26 x 109/L, Eosinophils 1.32 x 109/L, Platelet 656 x 109/L |
11.5 – 13.1 g/L 1.0 – 7.0 109/L 6.0 – 16.0 x 109/L 0.1 – 1.0 x 109/L 150 – 410 x 109/L |
| Immunoglobulin (Ig) |
IgG 2.14 IgA <0.06 IgM 0.058 IgE <1.0 |
2.50 – 6.80 g/L 0.01 – 0.06 g/L 0.06 – 0.21 g/L 1.0 – 29.0 IU/mL |
| T cells (absolute count) |
CD3 774 CD4 250 CD8 150 |
1767 – 3292 92 – 2289 726 – 1490 |
| Naive and Activated T cells (%) |
CD3+ CD45RA 27.1 CD3+ CD45RO 69.5 CD4+ CD45RA 0 CD4+ CD45RO 100 CD8+ CD45RA 40.5 CD8+ CD45RO 53.6 | Not available |
| B cells (CD20) (absolute count) | 1573 | 685 – 1597 |
|
NK cells (CD56+16) (absolute count) | 50 | 267 – 752 |
| Interphase FISH using CEP X/Y probe |
250 nuclei: 222 XY, 28 XX 11.2% maternal (XX) cells | Not applicable |
| Metaphase chromosome analysis |
33 metaphase lymphocytes: 3 XY, 30 XX 90.9% maternal (XX) cells | Not applicable |
List of microbial samplings conducted
| Microbial samplings | Results |
|---|---|
| Nasal | Positive for rhinovirus/enterovirus |
| Ear | Positive with mixed bacterial growth including |
| Urine | Positive |
He was commenced on intravenous immunoglobulin replacement therapy (3 g) every four weeks, together with prophylactic anti-infective agents: aciclovir suspension (HSV prophylaxis), isoniazid + rifampicin suspension (BCG prophylaxis), fluconazole (antifungal prophylaxis), and co-trimoxazole suspension for Pneumocystis jirovecii as per protocol (Table 3) 5. The Enterococcus urinary tract infection was treated with intravenous piperacillin/tazobactam. In addition, ciprofloxacin ear drops, desonide lotion for the scalp and trunk, and Daktacort for the perineum were prescribed to address otitis and cutaneous manifestations. Throughout therapy, he remained haemodynamically stable, with mild temperature fluctuations (36.4–38.5 °C).
An IFN-γ ELISPOT assay was performed, and the patient’s T cells secreted IFN-γ at levels comparable with those of healthy controls. Results are expressed as spot-forming units per 10⁶ cells (SFU/10⁶ cells) (Figure 2).
Prophylaxis prescribed during treatment (5).
| Prophylaxis | Drug doses | Disease / Infection |
|---|---|---|
| Aciclovir suspension | 130 mg orally three times a day. | HSV |
| Fluconazole capsule | 50 mg orally every morning. | Fungal |
| Isoniazid tablet | 100 mg orally every morning. | BCG |
| Rifampicin suspension | 64 mg orally every morning. | BCG |
| Co-trimoxazole suspension (Trimethoprim) | 32 mg orally every night. |
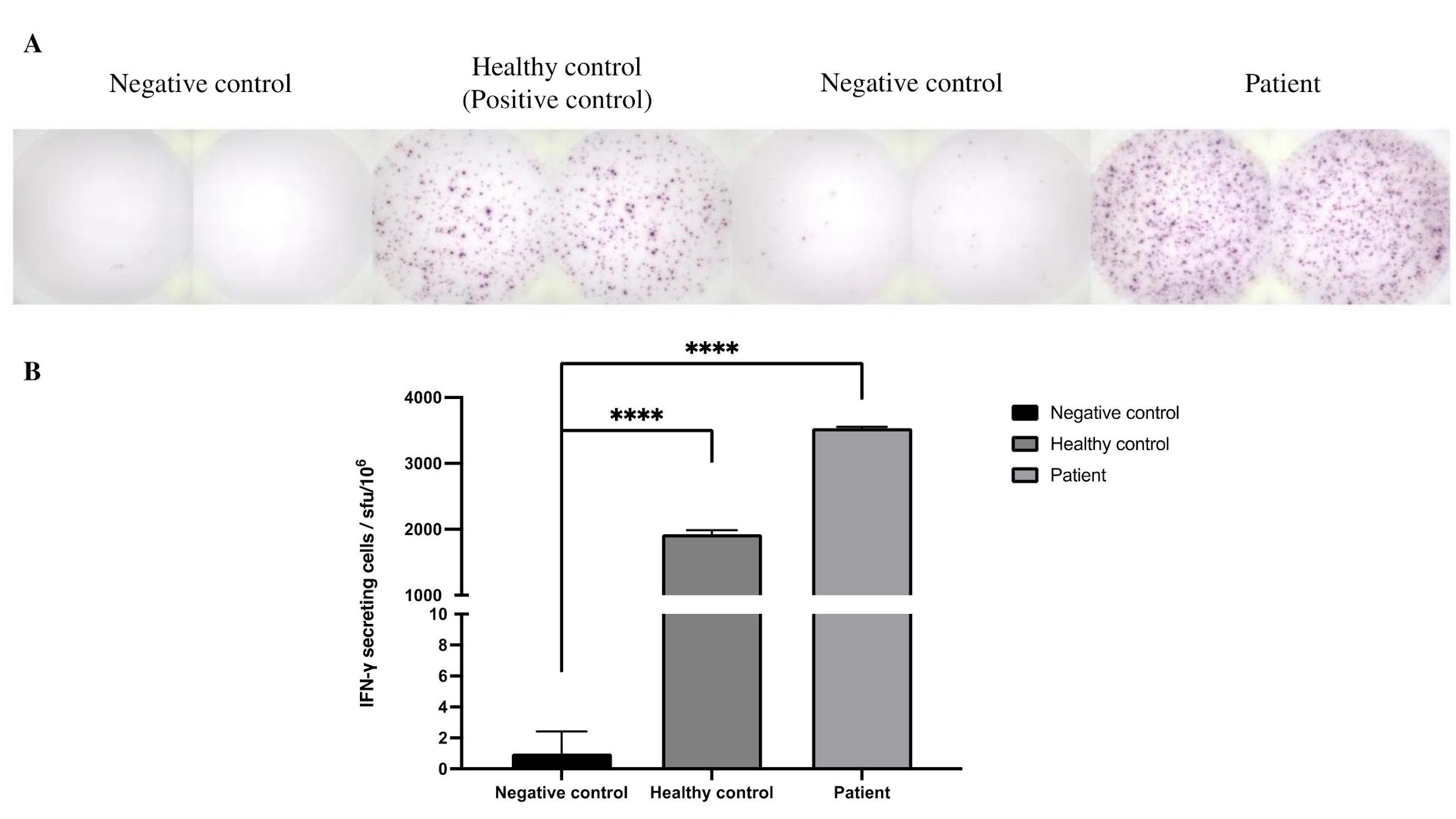
IFN-γ ELISPOT results. (a) Healthy control with average 1925 sfu/106 cells and patient with average 3535 sfu/106 cells. Peripheral blood mononuclear blood (PBMCs) from healthy control and patient were incubated overnight with mononuclear antibody, CD3-2 (anti-CD3 mAb), with 100,000 cells. Cells were removed and the plate was washed with PBS. Spots were counted using ImmunoSpot S6 Flex M2. (b) Spot counts of IFN-γ secreting cells. **** P < 0.0001 significantly different from negative control, with values represent the mean (±SD) of duplicate wells.
This finding further supports a diagnosis of leaky SCID with Omenn syndrome or SCID complicated by maternal T-cell engraftment (TME) and graft-versus-host disease (GVHD). Genetic testing confirmed X-linked SCID (X-SCID) with a likely pathogenic variant c.671G>C (p.Arg224Pro) in the IL2RG gene. Additionally, interphase fluorescence in situ hybridization (FISH) and metaphase chromosome analyses demonstrated maternal TME and GVHD, revealing 11.2 % and 90.9 % maternal cells, respectively (Table 1). The patient underwent an allogeneic haematopoietic stem-cell transplantation (HSCT) from an HLA-matched sister donor, using bone marrow as the stem-cell source. Conditioning consisted of rabbit antithymocyte globulin (ATG), fludarabine and treosulfan, followed by GVHD prophylaxis with sirolimus. A CD34+ cell dose of 6.4 × 10/kg was infused. Post-transplantation, the patient achieved full hematopoietic engraftment and immune reconstitution. At the most recent evaluation, the CD3+ T-cell count was 3,983 /µL, the CD19+ B-cell count 1,600 /µL, and the serum IgG level had normalised to 827 mg/dL, indicating adequate immune recovery. He remains clinically stable and is doing well.
DISCUSSION
First described in 1993, X-linked severe combined immunodeficiency (X-SCID) is the most common SCID subtype and is caused by pathogenic variants in the IL2RG gene. It accounts for approximately half of all reported SCID cases, with an incidence of 1:130,000 1,6. To date, 147 pathogenic mutations in the IL2RG gene have been characterised, with missense changes being the most frequent, followed by nonsense and frameshift variants 6. In Malaysia, SCID is the most frequently reported inborn error of immunity (IEI), with X-SCID identified in three of seven genetically confirmed SCID patients 7. Without definitive therapy—such as haematopoietic stem-cell transplantation (HSCT) or gene therapy—mortality within the first two years of life is common 3,8. Among X-SCID patients, laboratory evaluation typically demonstrates normal B-cell counts, markedly reduced T and natural killer (NK) cells, a virtual absence of naïve CD45RA⁺ T cells, and decreased memory CD45RO⁺ T cells. Functional assays usually reveal severely impaired T-cell responses to mitogens (e.g., <10 % of normal phytohaemagglutinin-stimulated proliferation) and/or to anti-CD3 antibodies 3.
The γc/CD132 receptor subunit, encoded by the IL2RG gene, is crucial for immune function as a common signaling component of the interleukin (IL)-2, IL-4, IL-9, IL-15, and IL-21 receptor complexes, mediating the regulation of T-cell development, regulatory T-cell (Treg) function, NK-cell maturation, as well as B-cell activity 9,10.
IFN-γ, a type II interferon primarily secreted by activated T helper 1 (Th1) cells and activated CD8⁺ T cells, augments immune responses by inducing adaptive immune pathways that promote cellular proliferation and differentiation, as well as antiviral and antimicrobial functions 11.
Between 2014 and 2021, the clinical presentation and diagnostic landscape of severe combined immunodeficiency (SCID) changed with the implementation of newborn screening (NBS) based on quantification of T-cell receptor excision circles (TRECs) in dried blood spots 12. In 2023, the Primary Immune Deficiency Treatment Consortium (PIDTC) revised the diagnostic criteria for SCID to encompass typical SCID, leaky SCID, and Omenn syndrome 12.
In countries lacking NBS programs, SCID and leaky SCID are diagnosed mainly on the basis of clinical manifestations arising during early infancy. Affected infants frequently present with recurrent or persistent infections caused by opportunistic pathogens; failure to thrive; chronic diarrhoea; respiratory infections leading to bronchiectasis; warts; dermatitis; immune dysregulation; and autoimmunity 3,12. Early administration of intravenous immunoglobulin (IVIG) can promote partial or complete immune reconstitution in these patients 13.
In our case, although the patient lacked naïve CD4+CD45RA+ T cells, ELISPOT analysis demonstrated that these cells remained functional and were able to secrete IFN-γ, indicating preserved cell-signalling pathways involved in the immune response. Furthermore, the elevated levels of IFN-γ observed in the patient may reflect an active infection and/or excessive T-cell activation 12. Notably, cytogenetic analyses using fluorescence in situ hybridisation (FISH) and karyotyping supported the presence of maternal T cells. Using a CEP X/Y probe for interphase FISH, 11.2 % of T-lineage cells were of maternal origin, whereas metaphase chromosome analysis showed that 90.9 % of dividing T cells were maternal. Taken together, these findings suggest that the elevated IFN-γ could be attributed to activation of the maternal engrafted T cells, further supporting the diagnosis of transmaternal engraftment (TME) 8,10,14. Additionally, erythroderma and chronic diarrhoea were considered manifestations of graft-versus-host disease (GVHD).
Prior to genetic testing, the case met all diagnostic criteria for X-linked severe combined immunodeficiency (X-SCID). According to the PIDTC 2022 definitions, evidence was insufficient to categorise the patient as having Omenn syndrome (Table 4). Clinically, he experienced multiple, recurrent viral and opportunistic bacterial infections of the upper respiratory and urinary tracts, together with erythroderma and chronic diarrhoea.
Differences between patient’s maternal T-cell engraftment and Omenn syndrome based on PIDTC 2022 Definitions.
| Feature | Omenn Syndrome | Maternal T-Cell Engraftment (TME) / GVHD | Patient’s Findings & Interpretation |
|---|---|---|---|
| Genetic / T-cell origin |
Pathogenic gene variant (s) |
Absence of functional host T cells Engraftment of maternal T cells causing GVHD |
IL2RG pathogenic variant (X-SCID) Maternal chimerism (11.2% & 90.9%) → TME confirmed |
| T-cell phenotype |
Oligoclonal T cells >80% CD45RO⁺ memory Absent naïve CD4⁺ |
Variable T-cell count Maternal T cells = activated/memory Host naïve T cells absent |
T, NK lymphopenia No naïve CD4⁺ |
| Skin / rash |
Generalized erythroderma, desquamation and absence of TME |
GVHD-like erythroderma, desquamation due to maternal T cells |
Erythroderma since 3 weeks, peeling & thick scales |
| Lymphoid / organomegaly |
Lymphadenopathy, hepatosplenomegaly common |
Often absent or mild |
None detected → favours TME |
| Maternal chimerism |
Absent |
Present |
FISH: 11.2% & 90.9% maternal cells → diagnostic for TME |
Genetic analysis revealed a hemizygous, likely-pathogenic variant, c.671G>C (p.Arg224Pro), in exon 5 of the IL2RG gene, thereby confirming the diagnosis of X-linked severe combined immunodeficiency (SCID). This novel single-nucleotide variant substitutes the basic, polar arginine at position 224 with a neutral, non-polar proline in the IL2RG protein. Proline-224 is clinically relevant, as evidenced by other pathogenic substitutions at this residue (PMID: 9049783, 9058718, 9633906, 10792291, 21184155, 28747913). The most thoroughly documented pathogenic change is c.670C>T (p.Arg224Trp). Multiple publications 15 have reported p.Arg224Trp in patients with X-linked SCID, demonstrating aberrant signaling consistent with loss of γc-chain function, and underscoring that the Arg224 residue—and, by extension, Pro224—lies within a functionally critical region of IL2RG. The novel p.Arg224Pro variant eliminates a positively charged side chain and introduces a conformationally restrictive residue; proline frequently disrupts loops and reduces flexibility required for proper folding or receptor interaction. Although p.Arg224Pro is absent from population databases and unreported in the literature, in-silico structural modeling predicts a disruptive effect on IL2RG function with an 80 % positive predictive value. This variant has been submitted to ClinVar (ID 2768005) as likely pathogenic on the basis of cumulative clinical, computational, and segregation evidence.
During pregnancy, maternal cells can cross the partially bidirectional placental barrier and enter the fetus. Up to 40 % of infants with SCID, unlike immunocompetent newborns, are unable to eliminate these maternal cells, permitting alloreactive lymphocytes to attack host tissues and precipitate GVHD 16. GVHD secondary to TME may mimic the clinical and laboratory manifestations of Omenn syndrome, including erythroderma, eosinophilia, and dysregulated T-cell function. Nevertheless, Omenn syndrome generally exhibits a more-severe phenotype and is frequently associated with hypomorphic mutations in SCID-related genes such as RAG1 and RAG2 17. In our patient, the absence of metaphase male lymphocytes indicated impaired T-cell differentiation in the thymus attributable to γc deficiency. ELISPOT analysis demonstrated a high frequency of reactive T cells, supporting the diagnosis of GVHD secondary to TME.
CONCLUSION
Delay in diagnosis or initiation of treatment in X-linked severe combined immunodeficiency (X-SCID) is associated with a significantly higher mortality. Clinicians should maintain a high index of suspicion in patients who present with recurrent infections, heightened susceptibility to opportunistic pathogens, failure to thrive, chronic diarrhoea, or clinical manifestations suggestive of Omenn syndrome 3. Immunological investigations, including T-, B- and NK-cell enumeration (TBNK), fluorescence in situ hybridisation (FISH), and functional assessment by IFN-γ ELISPOT assay, allow rapid evaluation of T-cell competence. These assays may also aid in differentiating leaky SCID from T-cell immune dysregulation such as TME in suspected cases while confirmatory genetic testing is pending. Haematopoietic stem-cell transplantation (HSCT) markedly improves survival and quality of life in X-SCID, and longitudinal monitoring of post-transplant immune reconstitution with the IFN-γ ELISPOT assay is recommended.
ABBREVIATIONS
ATG: anti-thymocyte globulin; BCG: Bacillus Calmette-Guerin; CD: cluster of differentiation; FBC: full blood count; FISH: Fluorescent in-situ Hybridization; γc: common gamma chain; GVHD: graft versus host disease; HSCT: hematopoietic stem cell transplant; HSV: Herpes simplex virus; IEI: inborn error of immunity; IFN-γ ELISPOT: interferon gamma enzyme-linked immunosorbent spot; Ig: immunoglobulin; IL: interleukin; IL2RG: Interleukin 2 receptor subunit gamma; IVIG: intravenous immunoglobulin; NBS: newborn screening; NK: natural killer; PBMC: peripheral blood mononuclear cells; PIDTC: Primary Immune Deficiency Treatment Consortium; Th1: helper T-cell 1; TME: transplacental acquired maternal engraftment; TREC: T-cell receptor excision circle; TREG: Regulatory T cell; WHO: World Health Organization; X-SCID: X-linked Severe Combined Immunodeficiency.
ACKNOWLEDGEMENTS
The authors would like to thank the patient, his parents, Hospital Pakar Kanak-Kanak UKM, and KK Women’s and Children’s Hospital.
AUTHORS’ CONTRIBUTIONS
Writing-original draft: NFZF, AA, ZTZ/ Writing-review & editing for important intellectual content: AA, IJAH, ZTZ. All authors read and approved the final manuscript.
FUNDING
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Data and materials used and/or analysed during the current study are available from the corresponding author on reasonable request.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Written informed consent was obtained from the patient’s parents for publication of this Case Report and any accompanying images. Ethical approval was obtained from UKM Research Ethics Secretariat with ethics reference number JEP-2024-1000. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
COMPETING INTERESTS
The authors declare that they have no competing interests.