

Downregulation of choline kinase alpha by mir-32-5p promotes apoptosis and reduces migration in hepatocellular carcinoma cells
- School of Health Sciences, Health Campus, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia
- Nanotechnology in Veterinary Medicine Research Group, Faculty of Veterinary Medicine, Universiti Malaysia Kelantan, Pengkalan Chepa, Kelantan 16100 Malaysia
Abstract
Introduction: Hepatocellular carcinoma (HCC) remains one of the most challenging malignancies worldwide, owing to its increasing incidence and the limited effectiveness of current therapies. Elucidating the molecular mechanisms that drive HCC tumorigenesis is critical for identifying novel therapeutic targets. This study evaluates the role of microRNA-32-5p (miR-32-5p) in the post-transcriptional regulation of choline kinase alpha (chka), an oncogene implicated in several cancers, including HCC.
Methods: HepG2 human HCC cells were used as the experimental model to examine the interaction between miR-32-5p and chka. Dual-luciferase reporter assays confirmed direct binding to the chka 3′-untranslated region (3′-UTR), whereas quantitative real-time PCR quantified transcript abundance. Functional consequences were assessed using Annexin V apoptosis assays and scratch-wound migration assays. In-silico analysis predicted a miR-32-5p binding site within the chka 3′-UTR, which was subsequently validated by reporter assays demonstrating miR-32-5p-dependent chka down-regulation.
Results: Transfection of miR-32-5p mimics into HepG2 cells significantly reduced chka mRNA levels, induced apoptosis, and impaired cell migration. These findings indicate that miR-32-5p exerts a tumor-suppressive effect in HCC, at least in part through chka repression.
Conclusion: The present study strengthens the evidence that miRNAs are critical modulators of HCC pathogenesis and highlights the miR-32-5p/chka axis as a promising therapeutic target, particularly for tumors refractory to existing treatments.
Introduction
Among the most impactful cancers worldwide, hepatocellular carcinoma (HCC) is characterized by its high incidence and substantial contribution to cancer-related mortality. Over the past two decades, its prevalence has continued to increase and is projected to rise further in certain regions by 2030 1. HCC is especially common in Asia and Africa, where the high prevalence of hepatitis B virus (HBV) and hepatitis C virus (HCV) infections promotes the development of chronic liver disease and, consequently, HCC. Although liver transplantation remains the most effective treatment for HCC, the scarcity of suitable deceased-donor organs necessitates alternative therapeutic approaches. These alternatives include surgical resection, radiofrequency ablation, and systemic therapies used either as a bridge to transplantation or to delay recurrence 2. Currently, the following agents are approved for first-line treatment of unresectable HCC: sorafenib, lenvatinib, atezolizumab plus bevacizumab, and tremelimumab plus durvalumab. The tremelimumab–durvalumab combination has been approved for first-line treatment of unresectable HCC in the United States 3 and Japan 4. Nonetheless, acquired resistance to these regimens has been documented 5.
Carcinogenesis proceeds through a series of genetic alterations that disrupt the normal regulation of essential cellular processes 6. Recognition that specific genes can modulate signal-transduction pathways has driven efforts to identify novel oncogenes and clarify their roles in tumour initiation and progression. Such identification is fundamental to the development of targeted anticancer strategies aimed at improving therapeutic efficacy. One oncogene of interest is choline kinase α (chka), which plays a pivotal role in tumorigenesis. Overexpression of chka has been documented as a common clinical feature in multiple malignancies, including prostate 7, ovarian 8, breast 9, and hepatic 10 cancers.
Choline kinase (CHK; EC 2.7.1.32) catalyzes the first committed step of the CDP-choline (Kennedy) pathway, which is essential for the biosynthesis of phosphatidylcholine (PC), the predominant phospholipid of eukaryotic membranes. The enzyme uses ATP to phosphorylate free choline, thereby generating phosphocholine. In mammalian cells, chka is expressed as three cytosolic isoforms encoded by two distinct genes: chka, located on chromosome 11q13.1, and chkb, on chromosome 22q13.33. The chkb gene encodes a single 43 kDa isoform, whereas chka produces two splice variants—CHKA1 (52 kDa) and CHKA2 (50 kDa)—that differ by an 18-amino-acid insert. All isoforms form homo- or heterodimeric assemblies, and the crystal structures of the homodimers of CHKA and CHKB have been solved 11.
Given the well-documented oncogenic activity of chka, the α-isoform is now regarded as a robust biomarker for cancer diagnosis and prognosis. Pharmacological inhibition of CHKA exerts potent anticancer effects in vitro and in vivo12. The prototypical inhibitor hemicholinium-3 (HC-3) paved the way for second-generation inhibitors such as MN58b, RSM-932A, V-11-0711, and NMS-P830 13,14. In addition, RNA interference (RNAi)-mediated silencing of chka elicits comparable antiproliferative responses and induces apoptosis in multiple cancer cell lines while sparing normal cells 15. Here, we provide the first experimental evidence that miR-32-5p down-regulates chka in hepatocellular carcinoma, thereby extending the tumour-suppressive repertoire of this microRNA.
Human miR-32-5p (MiRBase accession MIMAT0000090) originates from the precursor pre-miR-32 and is transcribed from the MIR32 gene (NCBI NR_029506) located at 9q31.3. To the best of our knowledge, this is the inaugural report validating the direct interaction between hsa-miR-32-5p and the chka 3′-UTR via a dual-luciferase reporter assay in HepG2 cells, providing mechanistic insight into the post-transcriptional regulation of chka in hepatocellular carcinoma and corroborating prior in silico predictions.
Materials-Methods
Materials
The HepG2 human hepatocellular carcinoma cell line (ATCC HB-8065) was obtained from the American Type Culture Collection and was authenticated and verified to be mycoplasma-free. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific) supplemented with 10 % heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific). To prevent microbial contamination, the medium was additionally supplemented with penicillin (100 units mL) and streptomycin (100 µg mL) (both from Sigma-Aldrich, Merck KGaA). The cells were maintained in a humidified incubator at 37 °C with 5 % CO.
GE Healthcare Dharmacon supplied the miRIDIAN microRNA mimic Housekeeping Positive Control #2, which targets GAPDH, along with the miRIDIAN Negative Control #1. Applied Biological Materials provided the synthetic hsa-miR-32-5p mimic (5′-UAUUGCACAUUACUAAGUUGCA-3′) together with a sequence-specific inhibitor directed against the same miRNA. All microRNA reagents were reconstituted in 1 × siRNA buffer (GE Healthcare Dharmacon) according to the manufacturers’ instructions.
OriGene Technologies supplied the pMirTarget reporter plasmid containing the 3′-untranslated region (3′-UTR) of the CHKA gene linked to a firefly luciferase reporter.
Oligonucleotide Transfection
Lipofectamine 3000 (Thermo Fisher Scientific, Inc.) was used to transiently transfect adherent HepG2 cells with various nucleic acid constructs. These constructs comprised the hsa-miR-32-5p mimic, its sequence-specific inhibitor, corresponding negative-control oligonucleotides, and the pMirTarget-CHKA-3′-UTR luciferase reporter plasmid. MicroRNA target validation was performed in 96-well plates, whereas qRT-PCR and other functional assays were carried out in 24-well plates. The plates were seeded with 1 × 10 cells per well (96-well) or 1 × 10 cells per well (24-well) approximately 16–18 h before transfection, allowing cultures to reach 70–80 % confluence at the time of nucleic acid delivery.
For complex formation, the pMirTarget-CHKA-3′-UTR plasmid or empty vector was combined with the hsa-miR-32-5p mimic and/or its inhibitor in Opti-MEM Reduced Serum Medium. Final concentrations were 200 ng plasmid DNA and 25 nM for each miRNA component. This miRNA concentration is widely validated for efficacy with minimal cytotoxicity; future experiments will include dose-response and time-course analyses. Lipofectamine 3000 was diluted separately in Opti-MEM according to the manufacturer’s instructions, after which the two solutions were mixed and incubated for 20 min at room temperature to allow lipid–nucleic acid complex formation. Untransfected control cells received Lipofectamine alone to account for reagent-related effects; no pharmacological positive control for apoptosis (e.g., staurosporine) was included.
In 96-well plates, each well received equal volumes of transfection complex and complete DMEM to a final volume of 100 µL. After a 6-h incubation, the medium was replaced with fresh complete medium, and cells were incubated for an additional 24 h before luciferase measurement. In 24-well plates, the ratio of complete DMEM to transfection mixture was 4:1, giving a total volume of 500 µL; these cultures were harvested 48 h post-transfection for downstream analyses.
Firefly Luciferase Reporter Assay
To evaluate the miRNA-dependent regulation of CHKA, HepG2 cells were transfected with the pMirTarget-CHKA-3′-UTR reporter plasmid and co-treated with either an hsa-miR-32-5p mimic (5′-UAUUGCACAUUACUAAGUUGCA-3′), its corresponding inhibitor (Applied Biological Materials), or a non-targeting negative-control mimic (GE Healthcare Dharmacon), in accordance with the transfection protocol. According to the manufacturer, the miR-32-5p inhibitor exhibits efficient intracellular delivery; future experiments will verify cellular uptake by fluorescence labelling and quantitative PCR. Twenty-four hours after transfection, 25 µL of culture supernatant was removed from each well, 75 µL of Dual-Glo Luciferase Reagent (Promega) was added, and plates were incubated for 10 min at room temperature to allow signal development. Firefly luciferase activity was measured on a GloMax 20/20 luminometer (Promega) and reported as relative light units (RLU). Each experimental condition was assessed in triplicate wells, and the entire assay was independently repeated at least twice. Luminescence values were normalised to those generated by the negative-control mimic to account for non-specific effects.
RNA Extraction and qRT-PCR
HepG2 cells were transfected with various microRNA constructs, including the hsa-miR-32-5p mimic, its specific inhibitor, their combination, a non-targeting negative control (miRIDIAN), and a GAPDH-targeting positive control. After incubation under standard culture conditions, cells were harvested, and total RNA was isolated using the Total RNA Isolation Kit (Thermo Fisher Scientific, Inc.) in strict accordance with the manufacturer’s instructions to ensure RNA purity and integrity. To eliminate potential genomic DNA contamination, RNA samples were treated with the RNase-Free DNase Set (Qiagen GmbH) prior to subsequent analyses. The concentration and purity of the extracted RNA were assessed on a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Inc.), and samples displaying appropriate A260/A280 and A260/A230 ratios were retained for further processing. Complementary DNA (cDNA) was synthesized using the RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Inc.). Quantitative real-time PCR (qPCR) was performed on an ABI Prism 7500 system with SYBR Green chemistry under optimized gene-specific thermal cycling conditions. Relative gene expression levels were normalized to the geometric mean of YWHAZ and RPS18 Cq values and calculated with the 2^−ΔΔCq method. The entire procedure for cDNA synthesis and qRT-PCR was conducted as previously described 16.
Apoptotic and dead cell count
Apoptotic and dead cells were evaluated using the Muse Annexin V & Dead Cell Assay Kit (Merck Millipore) in conjunction with the Muse Cell Analyzer (EMD Millipore), in accordance with the protocol previously described 16. These cell populations were quantified according to the manufacturer’s standard operating procedure.
Scratch wound healing assay
The scratch-wound healing assay was performed in accordance with the protocol previously described by our group 16. Briefly, HepG2 cells transfected with miRNA mimics were subjected to a linear wound, and wound closure was monitored over time. Images were captured at designated intervals, and the migratory distance was quantified using ImageJ software (National Institutes of Health).
Statistical analysis
Statistical analyses were performed with Student’s t-test or one-way ANOVA, followed by Tukey’s Honestly Significant Difference (HSD) post-hoc test. Statistical significance was set at p < 0.05. The assumption of normality was confirmed using the Shapiro–Wilk test before applying parametric tests. Exact p-values are reported in the figures. All analyses were conducted with IBM SPSS Statistics, version 22.0. Effect sizes (η) and observed power were calculated in SPSS; an n = 3 design was deemed sufficient for exploratory studies. Unless otherwise specified, data are presented as the mean ± standard error of the mean (SEM) derived from three independent experimental replicates.
Results
Validation of miR-32-5p Targeting of chka mRNA
In silico tools, including TargetScan, DIANA Tools microT-CDS, microRNA.org and miRDB (all referencing miRBase Release 23, August 2014), predicted that miR-32-5p targets the 3' untranslated region (3'UTR) of CHKA mRNA. To experimentally confirm this prediction, a dual-luciferase reporter assay was performed in HepG2 cells. Co-transfection of miR-32-5p with the reporter construct resulted in a 54 % reduction in firefly luciferase activity, indicating that miR-32-5p directly binds to the 3'UTR of CHKA mRNA (Figure 1). Conversely, transfection with a miR-32-5p inhibitor significantly increased luciferase activity, thereby supporting the specificity of this interaction and its reversibility upon miRNA inhibition.
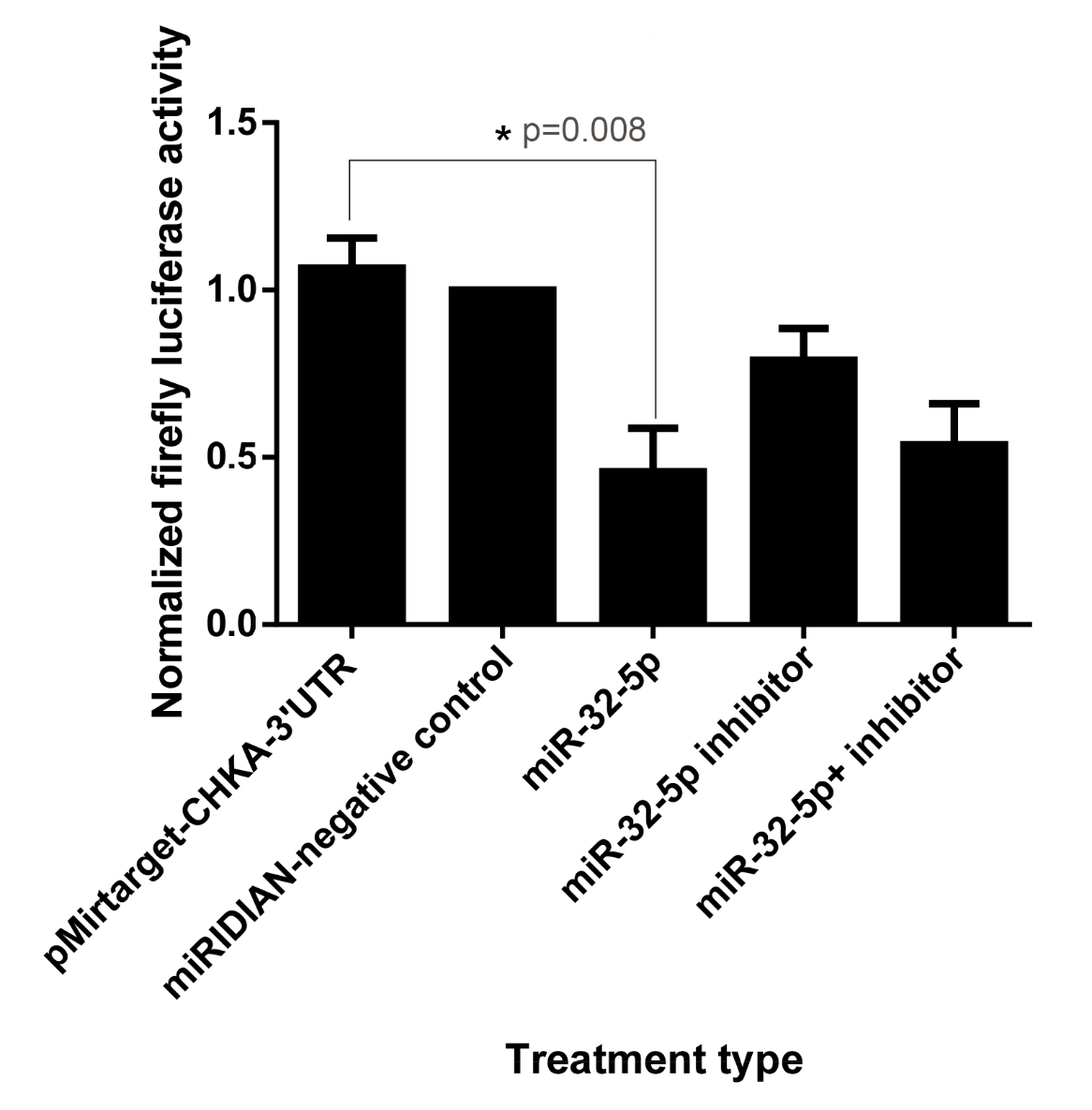
Target validation of miR-32-5p was performed by transfecting HepG2 cells with pMirTarget-chka-3'-UTR, followed by treatment with the specified miRNAs and miRNA inhibitors, either individually or in combination. Error bars represent the standard error of the mean (SEM) from triplicate experiments. Asterisk denotes a significant difference (One-way ANOVA with post-hoc Tukey HSD, p<0.05) when compared to untreated (no treatment) cells and those treated with miRIDIAN-negative control. The one-way ANOVA showed a statistically significant difference among the five groups (F(4,10) = 6.83, p = 0.0078). The large effect size (η2 = 0.732; Cohen’s f = 1.65) and high observed power (0.972) confirm that miR-32-5p significantly modulates CHKA 3′UTR-driven luciferase expression in HepG2 cells, consistent with strong post-transcriptional regulation.
miR-32-5p Suppresses Endogenous chka Expression in HepG2 Cells
To assess the regulatory effect of miR-32-5p on chka gene expression, HepG2 hepatoma cells were transfected with a synthetic miR-32-5p mimic. Transfection elicited a significant reduction in chka mRNA, decreasing by approximately 44% relative to both untreated controls and cells transfected with a scrambled negative control (Figure 2). Co-transfection of the miR-32-5p inhibitor with the mimic did not rescue chka expression, suggesting a robust and persistent silencing effect under the experimental conditions. These findings underscore the necessity for additional investigations into the interaction between miR-32-5p, its inhibitor, and the molecular mechanisms governing chka regulation.
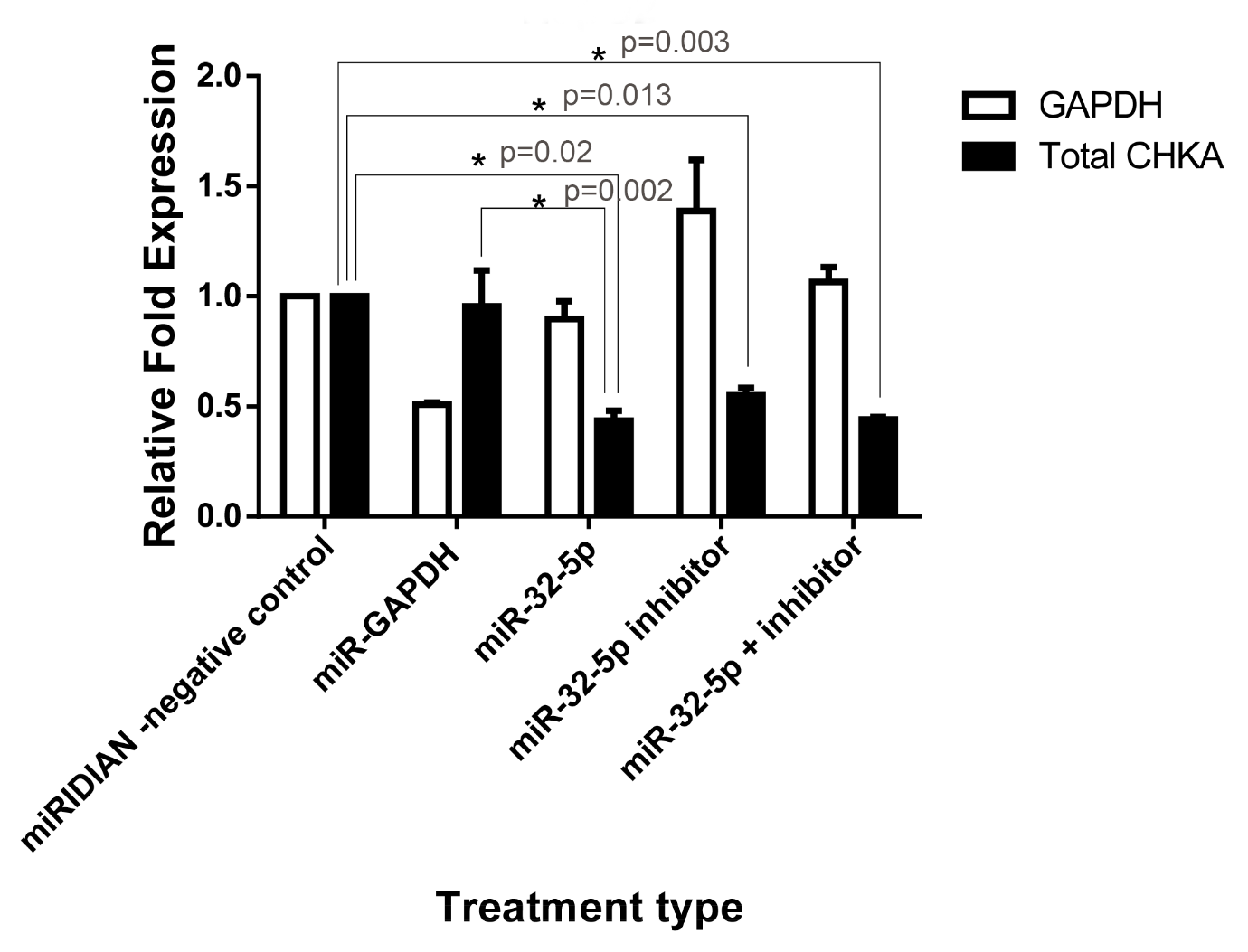
The impact of hsa-miR-32-5p mimic transfection on
miR-32-5p Induces Apoptosis in HepG2 Cells
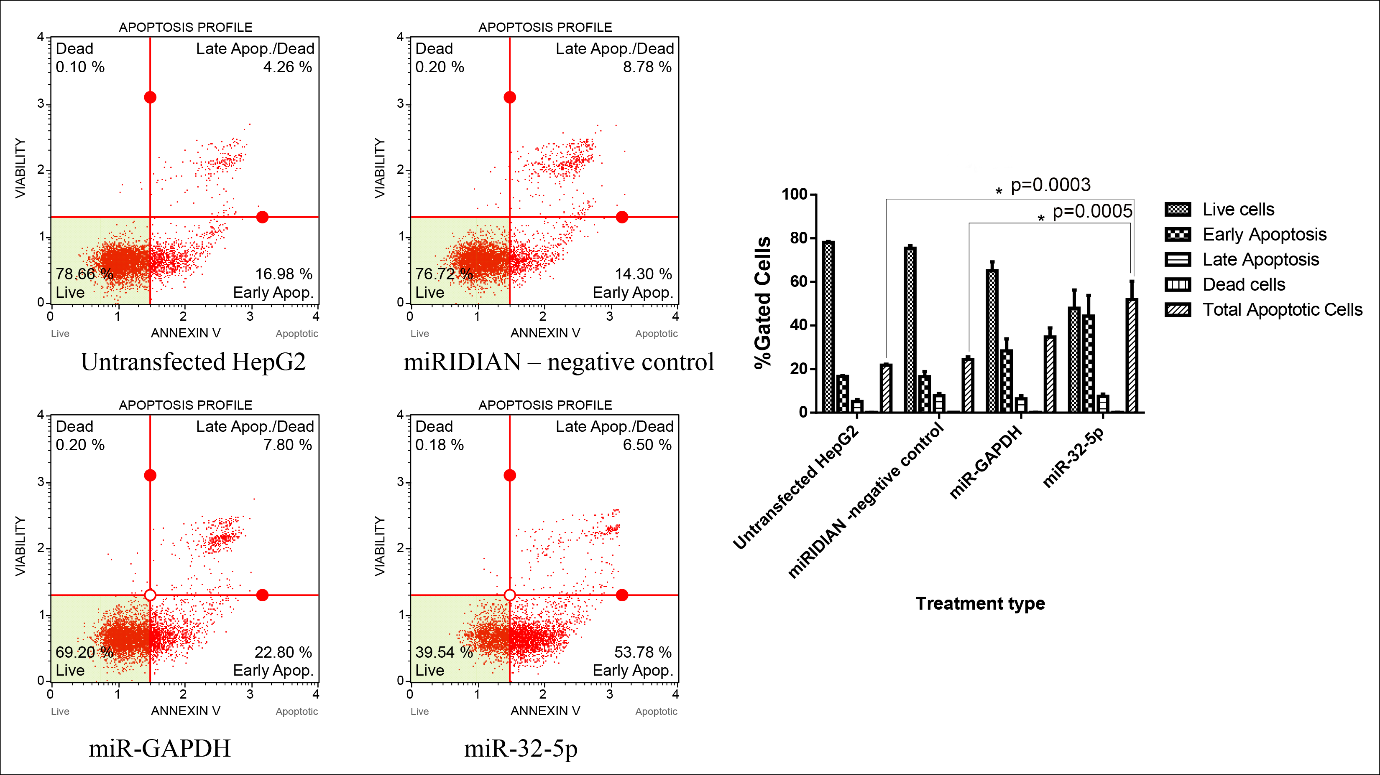
Previous studies have shown that inhibition of choline kinase α (CHKA) activity or RNA-interference-mediated silencing of CHKA expression induces apoptosis in various cancer cell lines 17. In light of these observations, we examined whether ectopic expression of an miR-32-5p mimic could elicit a comparable pro-apoptotic effect. Transfection with miR-32-5p significantly decreased cell viability and increased the proportion of Annexin V-positive cells relative to the negative-control miRNA. These results support a pro-apoptotic function of miR-32-5p, presumably through down-regulation of CHKA. Flow-cytometric analysis using Annexin V-FITC/propidium iodide staining revealed that the total apoptotic fraction rose to 51.94% after miR-32-5p transfection, compared with 24.37% in negative-control-transfected cells and 21.78% in untreated controls. Concordantly, the percentage of viable cells declined to 39.54% following miR-32-5p introduction, whereas 76.72% and 78.66% viability were recorded in the negative-control and untreated groups, respectively (Figure 3).
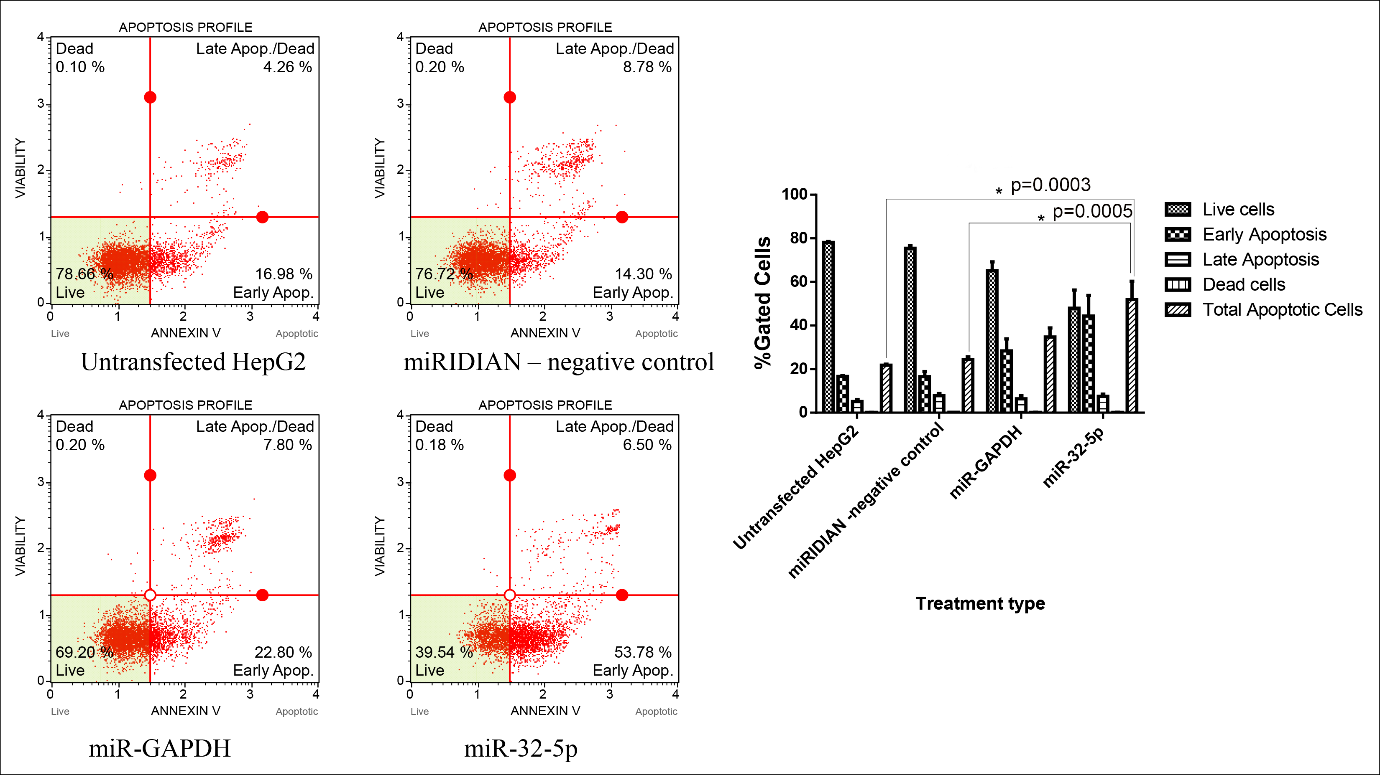
The effect of miR-32-5p on CHKA-regulated biological functions - cell apoptosis. miR-32-5p induced a significant increase in apoptosis in HepG2 cells. An asterisk denotes a significant difference. One-way ANOVA indicated a highly significant difference among the three groups (F(2,6)=42.93, p=0.00025, η2 = 0.935, Cohen’s f = 3.78, observed power = 0.999), demonstrating a strong treatment effect of the miR-32-5p mimic compared to controls.
miR-32-5p Impairs HepG2 Cell Migration
Scratch-wound assays demonstrated a significant decrease in the migratory capacity of HepG2 cells after transfection with miR-32-5p. At 72 h, miR-32-5p–transfected cells achieved approximately 40 % wound closure, whereas complete closure was observed in both untreated cells and cells transfected with the negative control (Figure 4).
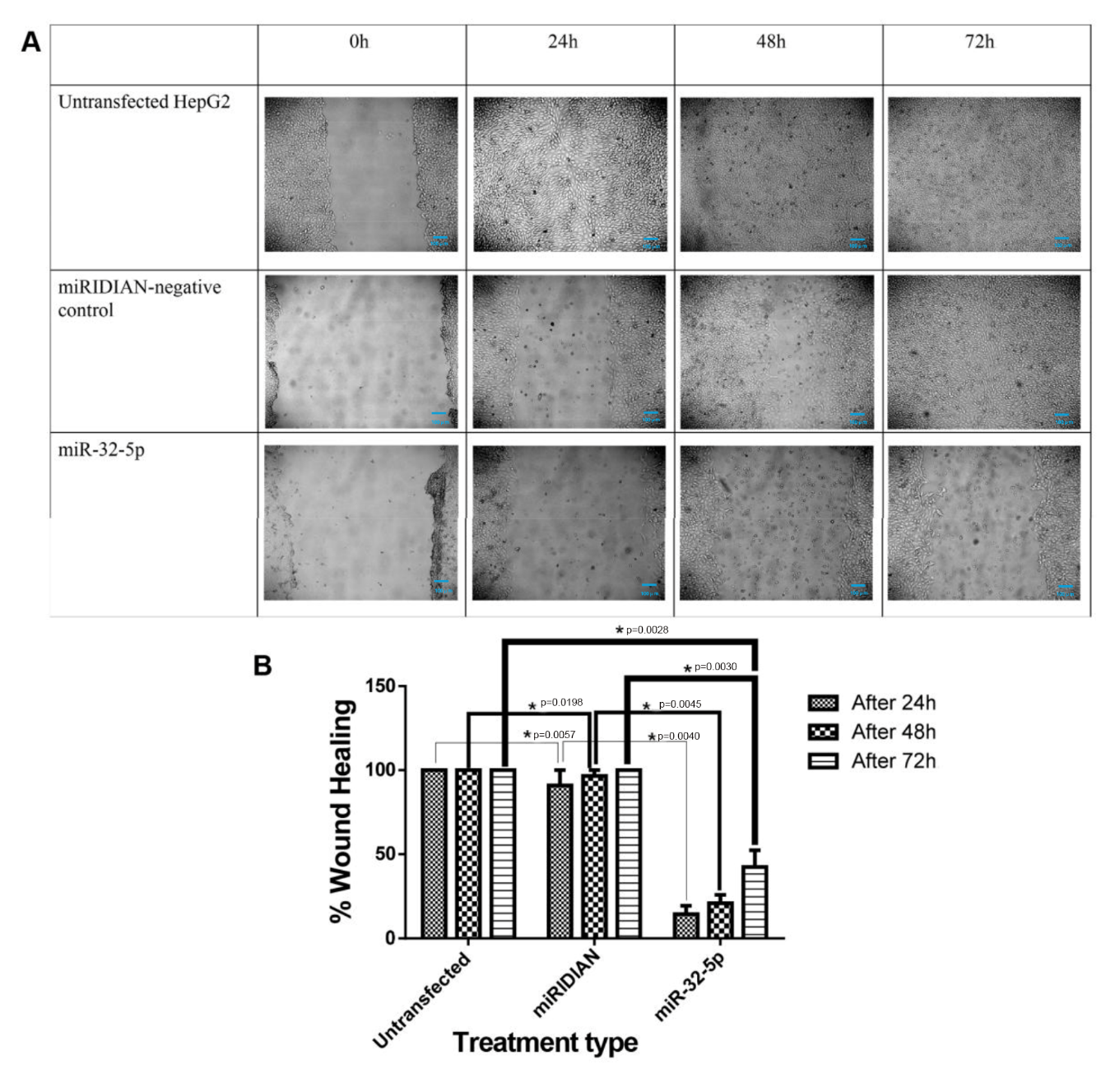
miR-32-5p inhibits cell migration in HepG2 cells with representative images of wound healing captured at 0-, 24-, 48-, and 72-hours post-scratch. Scale bar: 100 µm. The relative percentage of wound healing is presented. An asterisk indicates a significant difference at the respective time points when compared to untransfected cells and cells transfected with the negative control miRNA. Statistical analysis by one-way ANOVA revealed significant differences in cell viability among the three groups at each timepoint (24 h: F(2,6)=523.83, p<0.000001, η2=0.994, Cohen’s f = 13.21; 48 h: F(2,6)=917.10, p<0.000001, η2=0.997, Cohen’s f = 17.48; 72 h: F(2,6)=308.31, p=0.000001, η2=0.990, Cohen’s f = 10.14). Observed power = 1.000 for all analyses, indicating a very strong treatment effect.
Discussion
The results of this study provide strong evidence that miR-32-5p functions as a post-transcriptional inhibitor of CHKA expression in HepG2 liver cancer cells. Functional assays have demonstrated that overexpression of miR-32-5p leads to a marked decrease in CHKA mRNA levels, confirming a direct regulatory interaction. CHKA is a key enzyme involved in phospholipid synthesis. Aberrant upregulation of CHKA has been documented across numerous malignancies, including cancers of the liver, breast, prostate, and ovary. Its overexpression is frequently associated with enhanced tumour growth, increased metastatic potential, resistance to apoptosis, and poor clinical outcomes 18,19. Despite its well-established role in cancer biology, the molecular mechanisms driving CHKA overexpression remain incompletely defined. The current findings identify miR-32-5p as a previously unrecognized regulatory factor capable of suppressing CHKA expression at the post-transcriptional level. By reducing CHKA levels, miR-32-5p impairs tumour-promoting processes such as cell migration and cell survival, supporting its potential role as a tumour-suppressive miRNA. These insights contribute to a deeper understanding of the post-transcriptional regulation of CHKA and highlight miR-32-5p as a promising therapeutic candidate for the treatment of hepatocellular carcinoma. This work extends the miR-32-5p→CHKA axis to hepatocellular carcinoma, indicating that conserved post-transcriptional regulation exists across distinct malignancies.
The dual-luciferase reporter assay confirmed that miR-32-5p directly binds to the 3′ untranslated region (UTR) of chka mRNA, thereby corroborating in silico predictions of a regulatory interaction. This assay provides unequivocal evidence of target engagement and, at this exploratory stage, serves as a functional surrogate for rescue experiments. Although rescue studies would further demonstrate phenotypic restoration after miR-32-5p inhibition, the present luciferase assay directly validated the specificity of miR-32-5p binding to the chka 3′-UTR, a method widely regarded as the gold standard for establishing direct miRNA–target relationships because it isolates the post-transcriptional event from downstream signalling cascades. Consistent with a specific interaction, luciferase activity decreased significantly following miR-32-5p transfection and was restored by a miR-32-5p inhibitor. Collectively, these data indicate that miR-32-5p negatively regulates oncogenic chka expression and underscore its potential as a therapeutic lever against chka -driven tumorigenesis.
In functional assays, miR-32-5p reduced chka mRNA levels, promoted apoptosis, and suppressed migration in HepG2 cells. These effects mirror previous reports showing that genetic or pharmacological ablation of chka diminishes cancer cell proliferation and augments apoptosis in diverse tumour models 15. The inability to fully rescue chka expression after miR-32-5p inhibition may reflect the dominant suppressive capacity of the miRNA, suboptimal inhibitor potency, or rapid turnover of the target transcript, warranting further mechanistic investigation.
Furthermore, the diminished migratory capacity of HepG2 cells after miR-32-5p transfection aligns with existing evidence that CHKA activity facilitates tumour cell motility and metastatic behaviour 10,20. An approximately 60 % decrease in wound closure strongly suggests that miR-32-5p can modulate cytoskeletal organisation and migratory signalling, potentially through both CHKA-dependent and CHKA-independent mechanisms. Although scratch assays provide preliminary insight into migratory behaviour, future studies will employ trans-well assays and three-dimensional (3D) spheroid models for a more comprehensive assessment.
Collectively, our results highlight miR-32-5p as a promising target for therapeutic modulation in hepatocellular carcinoma. Analogous to other tumour-suppressive miRNAs, such as miR-145 and miR-370, which regulate oncogenic signalling pathways and cell fate 21,22, miR-32-5p contributes to a complex regulatory network with translational potential. This proof-of-concept study utilised HepG2 cells as a representative HCC model; future work will extend these observations to Huh7 cells and non-tumorigenic hepatocytes (THLE-2) to confirm cancer-specific effects.
Conclusions
In conclusion, the intricate interplay between miRNAs and gene expression in liver cancer provides crucial insights for the development of novel therapeutic approaches. Targeting specific miRNAs, such as miR-32-5p, which regulates the oncogene choline kinase α (chka), represents a promising strategy to modulate cancer progression. MicroRNA-based therapeutic strategies, owing to their ability to influence the molecular mechanisms that drive tumor initiation and progression, constitute a compelling avenue for improving liver cancer treatment and patient outcomes. Nevertheless, these observations warrant further in vivo validation.
Abbreviations
3D: three-dimensional; 3'UTR: 3' untranslated region; ANOVA: Analysis of Variance; ATP: Adenosine Triphosphate; CDP: Cytidine Diphosphate; cDNA: complementary DNA; CHK: Choline Kinase; chka: choline kinase alpha gene; CHKA1: Choline Kinase Alpha 1; CHKA2: Choline Kinase Alpha 2; CHKB: Choline Kinase Beta; DMEM: Dulbecco's Modified Eagle Medium; DNA: Deoxyribonucleic Acid; FBS: Fetal Bovine Serum; HCC: Hepatocellular Carcinoma; HC-3: Hemicholinium-3; HBV: Hepatitis B Virus; HCV: Hepatitis C Virus; HSD: Honestly Significant Difference; miRNA: microRNA; mRNA: messenger RNA; PC: Phosphatidylcholine; PCR: Polymerase Chain Reaction; qPCR: quantitative PCR; qRT-PCR: quantitative Real-Time PCR; RLU: Relative Light Units; RNA: Ribonucleic Acid; RNAi: RNA interference; SEM: Standard Error of the Mean
Acknowledgements
We extend our gratitude to the laboratory staff of School of Health Sciences, Universiti Sains Malaysia (USM), for their valuable technical support. We also wish to acknowledge the technical assistance provided by the Central Research Laboratory at the School of Medical Sciences, USM, as well as the Institute for Research in Molecular Medicine (INFORMM), Health Campus, USM.
Author’s contributions
The conception and design of the study were collaboratively developed by Wei Cun See Too, Ling Ling Few, Shuhaila Mat-Sharani, Nor Fadhilah Kamaruzzaman, and Get Bee Yvonne-Tee. Raikundalia Sweta was responsible for material preparation, as well as conducting data collection and analysis. Ling Ling Few and Wei Cun See Too prepared the initial draft of the manuscript. Funding acquisition was led by Wei Cun See Too, Shuhaila Mat-Sharani, Nor Fadhilah Kamaruzzaman, Get Bee Yvonne-Tee, and Ling Ling Few. All authors reviewed the manuscript critically for intellectual content and approved the final version for publication.
Funding
The work was supported by the Fundamental Research Grant Scheme (FRGS), provided by the Ministry of Higher Education Malaysia, under grant number FRGS/1/2024/SKK10/USM/02/7.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have not used generative AI (a type of artificial intelligence technology that can produce various types of content including text, imagery, audio and synthetic data. Examples include ChatGPT, NovelAI, Jasper AI, Rytr AI, DALL-E, etc) and AI-assisted technologies in the writing process before submission.
Competing interests
The authors declare that they have no competing interests.