

Amyloid Beta and Tau Aggregation: The Etiology and Potential Pharmaceutical Approaches for Alzheimer’s Disease
- Faculty of Pharmacy, Integral University, Dasauli, Kursi Road, Lucknow, Uttar Pradesh, 226026, India
Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized primarily by the accumulation of amyloid-β (Aβ) peptide and hyperphosphorylated, cleaved forms of the microtubule-associated protein tau. The probability of developing AD increases with age, mainly because the burdens of Aβ and tau pathology grow over time. Aβ plaques are composed of amyloid-β generated when β- and γ-secretases cleave the amyloid precursor protein (APP); these extracellular deposits disrupt neuronal homeostasis and ultimately trigger cell death. Neurofibrillary tangles formed by hyperphosphorylated tau compromise neuronal architecture and impair intracellular transport. This article discusses the formation of Aβ plaques and tau tangles as well as their potential modulation or clearance through interventions targeting molecules such as glycogen synthase kinase-3 (GSK-3) and fragment crystallizable receptors (FcRs). We also review the structures, mechanisms of action, neuropathological consequences, and synergistic effects of Aβ accumulation and tau phosphorylation. Monoclonal antibodies, including aducanumab and lecanemab, can slow plaque formation, neutralize Aβ toxicity, stimulate immune-mediated clearance, and remove existing aggregates. Tau-directed antibodies such as semorinemab and tilavonemab are currently in clinical trials and aim to lessen tau aggregation, stabilize microtubules, and inhibit pathogenic kinase activity. Advanced drug-delivery systems (e.g., exosome-loaded or peptide-conjugated nanoparticles) may facilitate the development of more precise, safer, and more potent therapeutics for AD.
Introduction
Alzheimer's disease is a neurodegenerative condition characterized by progressive forgetfulness, cognitive decline, impaired physical function, and ultimately death resulting from widespread neuronal loss1. Alzheimer’s disease (AD) is named after Dr. Alois Alzheimer, a German physician and pathologist who reported the first patient with the disorder in 19062. Alzheimer's disease is one of the greatest medical challenges of this century and the leading cause of dementia3. Globally, approximately 40 million people are estimated to have dementia, a figure projected to double roughly every two decades and to exceed 80 million by 20504. The prevalence of AD rises with age, increasing from about 27.6 % among individuals aged 65–74 years to roughly 36.4 % in those over 80 years5. AD pathology is characterized primarily by the formation of amyloid-β (Aβ) plaques and neurofibrillary tau tangles resulting from the accumulation of hyper-phosphorylated tau protein in the brain6. Aβ plaques are insoluble fibrillar structures composed of aggregated Aβ peptides in the extracellular space, whereas tau tangles are intracellular aggregates of abnormally phosphorylated tau protein that destabilize microtubule78. The interplay between these lesions is believed to drive the symptomatic progression of AD. Alzheimer's disease typically begins insidiously with difficulty remembering recent events and progresses gradually over time9. Acetylcholine (ACh), a neurotransmitter first isolated in 1867 and responsible for transmitting impulses between neurons as well as to voluntary and involuntary muscle cells, is found at reduced concentrations in the brains of individuals with AD10.
Methods
The present review utilized a range of scholarly search engines—Google Scholar, Semantic Scholar, ScienceOpen, and PubMed—as well as journal databases to identify recent primary and review articles. The search combined the keywords “Alzheimer’s disease,” “amyloid-β plaques,” “neurotoxicity,” “monoclonal antibodies,” “tau tangles,” “clinical trials,” and “neuroinflammation.” Inclusion criteria were English-language, peer-reviewed studies that addressed pathophysiology, pharmacological clinical trials, or drug-delivery advancements. In total, 138 articles published between 2011 and 2025 were selected for this review.
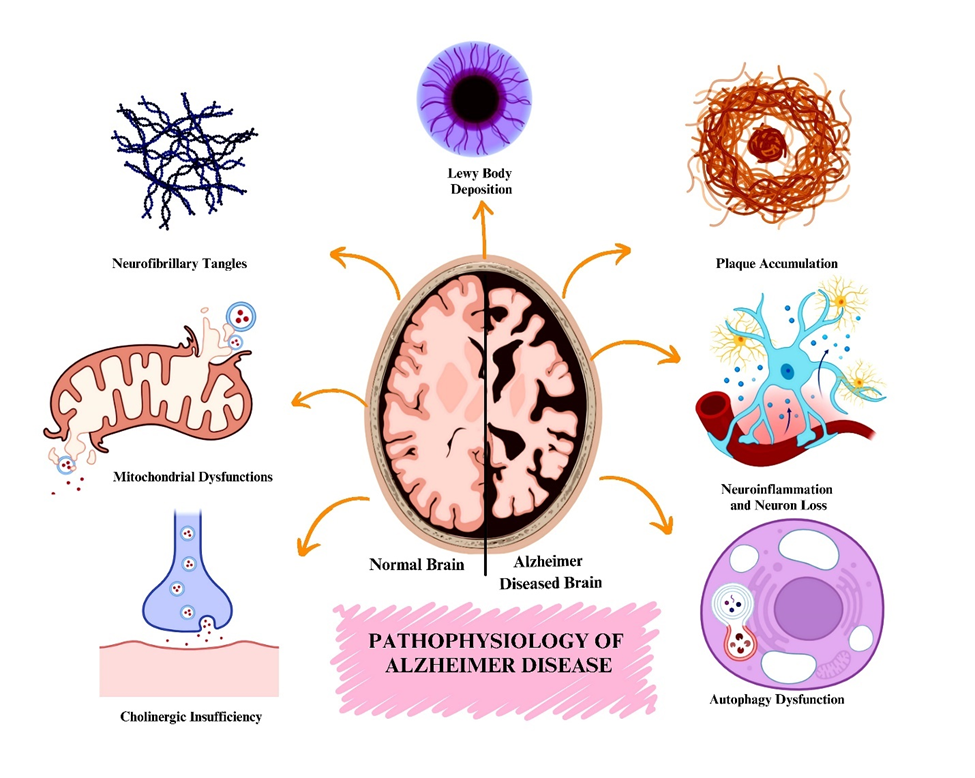
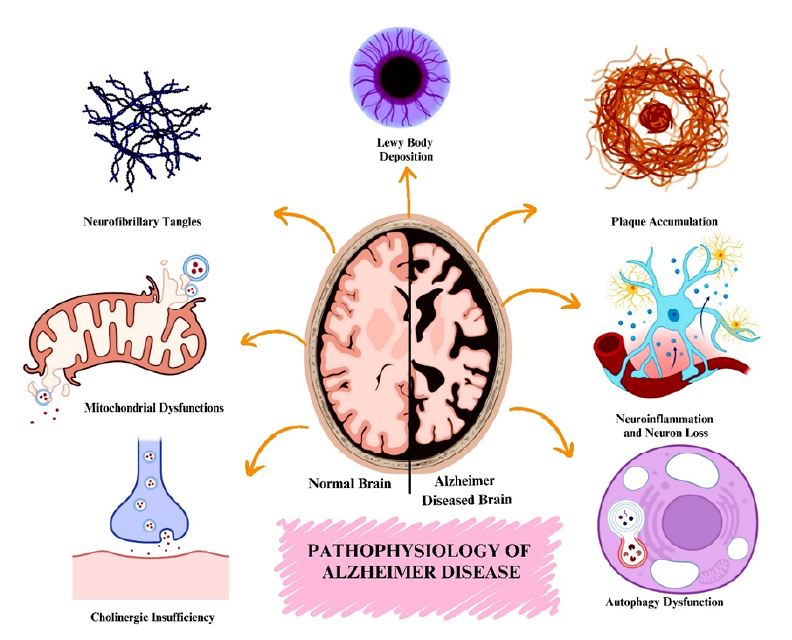
Pathophysiology of Alzheimer's Disease
AMYLOID β PLAQUES AND TAU TANGLES: THE CORE PATHOLOGY
Aβ plaques and tau tangles are now more thoroughly understood as pathologies of Alzheimer’s disease (AD). Both tau tangles and Aβ plaques are implicated in neurotoxic effects (Figure 11112). The interactions between Aβ and tau are major drivers of neurotoxicity, suggesting that these two AD pathological components synergistically increase neuronal damage. New phosphorylation sites of tau have been identified, and an understanding of tau seeding and spreading has deepened insight into tau pathology. These findings explain how tau spreads throughout neuronal networks and causes axonal degeneration in mammals13.
Aβ Plaques
Formation and Accumulation
Alzheimer’s disease is characterized by extracellular Aβ plaques whose precise pathogenesis remains incompletely understood. These plaques develop from the amyloid precursor protein (APP), which is cleaved by β- and γ-secretases. The resulting Aβ peptides are deposited in the brain, form insoluble plaques, impair cellular function, and ultimately drive neurodegeneration14.
Proteolytic Processing of APP
The processing of APP, a transmembrane glycoprotein expressed mainly in neurons, generates Aβ peptides. APP can follow either a non-amyloidogenic or an amyloidogenic pathway.
• Cleavage by β-Secretase (BACE1): APP is first cleaved by β-secretase, an aspartyl protease, in its extracellular domain. This cleavage produces two fragments: soluble APPβ (sAPPβ), which is released extracellularly, and the membrane-tethered C99 (β-CTF). C99 is the critical substrate for γ-secretase15.
• Cleavage by γ-Secretase: The multi-protein γ-secretase complex processes C99 within its transmembrane region, releasing Aβ peptides into the extracellular space and an intracellular APP domain (AICD). The most abundant isoforms are Aβ40 and Aβ4216. Aβ42 is highly aggregation-prone and constitutes the principal building block of amyloid plaques17.
Oligomerization of Aβ
Aβ peptides self-associate; Aβ42 aggregates more readily than Aβ40 because of its hydrophobic C-terminal end18. Aggregation proceeds from soluble monomers to toxic oligomers that disrupt neuronal signaling and synaptic plasticity, then to protofibrils, and finally to mature fibrils and plaques19.
Formation of Amyloid Fibrils
Protofibrils assemble into insoluble amyloid fibrils that form the structural core of plaques20. These fibrils adopt a characteristic cross-β-sheet conformation, conferring high stability and resistance to degradation2122.
Deposition and Plaque Development
Amyloid fibrils coalesce into extracellular deposits that constitute amyloid plaques, which are surrounded by dystrophic neurites, activated astrocytes and microglia, and extracellular matrix components such as apolipoprotein E (ApoE)2324.
Genetic mutations in APP, presenilin-1, or presenilin-2 (components of γ-secretase) increase Aβ42 production. The ApoE ε4 allele promotes aggregation and inhibits clearance. Impaired activity of Aβ-degrading enzymes (e.g., neprilysin) or reduced transport across the blood–brain barrier further enhances accumulation25.
Tau Tangles
Tau Phosphorylation and Aggregation
Tau is a microtubule-associated protein (MAP) that stabilizes microtubules in neurons. Hyperphosphorylated tau detaches from microtubules, aggregates, impairs axonal transport, and ultimately causes neuronal death13.
Formation and Function of Tau Protein
Tau is encoded by the MAPT gene on chromosome 17, which generates six isoforms via alternative splicing differing in their microtubule-binding repeats (3R vs. 4R) and N-terminal inserts. Native tau is intrinsically disordered, enabling dynamic interaction with microtubules26.
Pathological Transition of Tau
Under pathological conditions tau becomes hyperphosphorylated by kinases such as GSK-3β and CDK527. Detached tau undergoes conformational changes, forms soluble toxic oligomers, and then paired helical filaments (PHFs) and straight filaments (SFs). These filaments aggregate into neurofibrillary tangles (NFTs) that can propagate in a prion-like manner28.
Post-Translational Modifications (PTMs)
PTMs profoundly influence tau behavior. Hyperphosphorylation drives mislocalization and aggregation; acetylation hinders degradation; ubiquitination can target tau for proteasomal clearance or stabilize aggregates; truncation yields highly aggregation-prone fragments; and glycation further enhances aggregation propensity29.
Pathological tau disrupts microtubule integrity, axonal transport, synaptic plasticity, and mitochondrial function, and it induces neuroinflammation, all of which culminate in neuronal death30.
Tau pathology is promoted by MAPT mutations (e.g., P301L and the H1 haplotype)31, PTMs29, oxidative stress and neuroinflammation32, and an imbalance between kinases (e.g., GSK-3β) and phosphatases (e.g., PP2A)33.
Combined Impact on Cognitive Decline
The connection between Aβ and tau pathologies is robust. Whereas Aβ plaques contribute to early synaptic impairment and prodromal clinical symptoms, tau tangles correlate directly with neuronal loss and the severity of cognitive decline34.
Synergistic Effects
Aβ and tau pathologies are not independent; they interact sequentially and synergistically. Aβ plaques are thought to appear first, triggering cascades that lead to tau hyperphosphorylation and aggregation35.
MECHANISM OF ACTION OF Aβ AND TAU TARGETING DRUG
Aβ Targeting
Monoclonal antibodies (mAbs) are lab-engineered, Y-shaped protein structures36 composed of two heavy and two light chains. The variable region (Fab) at the ends binds to the target antigen37, while the constant region (Fc) interacts with immune-cell receptors38. The binding of mAbs to Aβ—in its soluble or plaque form—activates immune processes such as phagocytosis via Fc receptors on microglia, thereby facilitating plaque removal. Complement receptors also enhance clearance through the complement cascade3940. Although effective in promoting Aβ clearance, mAbs may induce inflammation through activation of Toll-like receptors (TLRs)4142. Drugs such as aducanumab exploit this mechanism to target Aβ and trigger microglial activation for plaque clearance, though their effects can be complex and require careful management43. Amyloid-related imaging abnormalities are serious side-effects of anti-Aβ antibodies (e.g., aducanumab, lecanemab, gantenerumab). Edema (ARIA-E) and hemosiderin-related hemorrhages (ARIA-H) are associated with disruption of the blood–brain barrier following amyloid clearance from cerebral vessels44. APOE-ε4 carriers face an increased risk. Symptoms include headaches, confusion, and seizures. Regular MRI monitoring is required for early detection, and dose adjustments may be necessary in severe cases45.
|
Aspect |
Aducanumab |
Lecanemab |
Gantenerumab |
|
Target |
Aggregated Aβ (Aβ) plaques and soluble oligomers |
Soluble Aβ (Aβ) protofibrils |
Aggregated Aβ (Aβ) plaques |
|
Mechanism of Action |
1. Binds aggregated Aβ plaques and oligomers. 2. Fc region activates microglia for plaque clearance. 3. Reduces amyloid burden, neuroinflammation, and slows cognitive decline |
1. Binds soluble Aβ protofibrils to prevent plaque formation. 2. Activates microglia to clear protofibrils and plaques. 3. Slows cognitive decline by reducing amyloid burden |
1. Binds aggregated Aβ plaques. 2. Disaggregates plaques and activates microglia for clearance. 3. Reduces amyloid burden and inhibits new plaque formation |
|
Efficacy |
Reduces amyloid burden and slows cognitive decline in early AD |
Delays cognitive and functional decline, with positive results in early AD |
Reduces amyloid plaques, but clinical efficacy in cognitive decline is still under study. |
|
Adverse Effects |
Amyloid related imaging abnormalities (ARIA), including brain swelling (ARIA-E) and microhemorrhages (ARIA-H) |
ARIA was observed but at a lower incidence compared to Aducanumab |
ARIA-E and ARIA-H, but typically at a lower incidence than Aducanumab |
|
Novelty |
Focused on plaque clearance in early-stage AD |
Targets protofibrils, a neurotoxic intermediate form of Aβ |
Subcutaneous delivery, potentially improving patient compliance |
Aducanumab
Aducanumab is a monoclonal antibody that targets Aβ and reduces plaque accumulation in patients with Alzheimer’s disease (AD) (Table 1)46. Several clinical studies show that aducanumab can slow cognitive decline in patients at an early stage of AD52. The aducanumab controversy was sparked by conflicting Phase 3 trial results (ENGAGE vs. EMERGE) and differing FDA-review interpretations. ENGAGE did not reach its primary endpoint, whereas EMERGE showed a modest slowing of cognitive decline53.
Lecanemab
Lecanemab is a monoclonal antibody directed against soluble Aβ protofibrils; it decreases amyloid plaques and clinical manifestations in early AD54. By neutralizing protofibrils, lecanemab reduces new-plaque formation and maintains a lower amyloid burden47. Collectively, trial data indicate that lecanemab delays both cognitive and functional decline. Study NCT03887455 (Table 2) showed that lecanemab significantly slowed clinical decline in early AD (CDR-SB; p < 0.0001)55.
|
Drug |
NCT Number |
Trial Phase / Duration |
Limitations |
Controversies |
Statistical Outcomes |
|
Aducanumab |
NCT01397539 |
Phase 1 (2011–2013) 53 participants |
Small sample, single-dose, short follow-up; efficacy assessment not possible. |
ARIA-E (100% in 60 mg/kg group); early safety concern but limited controversy due to phase and size. |
Dose-dependent ARIA-E (100% at 60 mg/kg); linear PK up to 30 mg/kg; cognition not statistically significant. |
|
NCT01677572 |
Phase 1b (2012–2019) 197 participants |
Small sample; short follow-up; focused on amyloid reduction not efficacy. |
Mild concern; no major controversy. |
Significant ( | |
|
NCT02477800 (EMERGE) |
Phase 3 (2015–2019) ~1,600 participants |
Early termination; inconsistent outcomes; post-hoc analysis; potential unblinding due to ARIA. |
FDA approval despite advisory panel rejection; EMERGE positive, ENGAGE negative; resignations from FDA board. |
22% decline reduction (CDR-SB −0.39; | |
|
NCT02484547 (ENGAGE) |
Phase 3 (2015–2019) ~1,600 participants |
Same as EMERGE; dose modification; no significant effect. |
Same as EMERGE; |
No significant effect; inconsistent with EMERGE. | |
|
NCT04241068 |
Phase 3 Extension (2020–2023) 2,400 participants |
No control group; limited to 10 mg/kg; single dose reporting; unclear participant number. |
Continued ARIA concern; treatment discontinuations due to adverse events; ADA positivity raised tolerability issues. |
Detailed statistical outcomes not available due to study termination. | |
|
Lecanemab |
NCT03887455 |
Phase 3 (Clarity AD, 2019–2022) 1,795 participants |
Targeted only early AD; amyloid confirmation required; generalizability limited. |
Debate over clinical significance of small CDR-SB change (0.45); ARIA-E (12.6%), ARIA-H (17.3%) despite mild severity. |
Highly significant ( |
|
Gantenerumab |
NCT03444870 |
Phase 3 (GRADUATE I/II, 2018–2023) 1,053 participants |
Failed primary endpoint (CDR-SB); no significant cognitive benefit; modest trends in secondary outcomes. |
Focused on amyloid reduction despite failed cognitive outcomes; subcutaneous route praised for compliance but raised concerns. |
Non-significant CDR-SB difference (−0.31; 95% CI: −0.66 to 0.05); significant amyloid reduction; ARIA-E observed in notable proportion. |
Gantenerumab
Gantenerumab is an antibody that binds aggregated Aβ and facilitates its removal from the brain48. Emerging studies suggest that gantenerumab reduces amyloid plaques and improves cognition. Although its mechanism resembles that of aducanumab, gantenerumab is delivered subcutaneously, a route that may enhance treatment adherence if approved1264. Ongoing trials are evaluating its efficacy, safety, and potential to slow early AD progression.
Tau-Targeting
Therapies that target tau tangles modulate several receptors and molecular partners. One key target is the microtubule-associated protein tau65. Glycogen synthase kinase-3 (GSK-3) inhibitors and mitogen-activated protein kinase (MAPK) inhibitors both reduce tau phosphorylation66. Because MAPKs mediate stress- and inflammation-related signaling, their down-regulation lessens phosphorylation and inflammation. These interactions decrease tau aggregation, stabilize microtubules, and reduce tau-induced neurotoxicity, thereby helping to maintain neuronal function and limiting tau’s prion-like propagation67.
Tau antisense oligonucleotides (ASOs)
Tau ASOs are short synthetic single-stranded RNAs or DNAs designed to bind a specific sequence within tau (MAPT) mRNA, thereby reducing tau translation68.
Mechanism of Action: Tau ASOs hybridize via Watson–Crick base pairing with tau mRNA, forming an RNA–DNA duplex that recruits RNase H, which cleaves the RNA strand69. RNase H–mediated cleavage is catalytic, so multiple mRNAs can be degraded. Lower tau levels limit formation of hyperphosphorylated tau, paired helical filaments, and neurofibrillary tangles70. Reduced aggregates lessen axonal transport disruption and neuronal dysfunction71.
IONIS-MAPTRx, delivered intrathecally, has shown significant reductions in cerebrospinal-fluid tau levels in early trials72.
Anti-tau antibodies
Anti-tau antibodies are therapeutic mAbs that bind pathological tau conformations in AD48. They are in various clinical and preclinical stages73.
Biomarker validation in AD still faces challenges of specificity, sensitivity, and clinical utility. Reliable biomarkers (Aβ, tau, NfL) must distinguish AD from other neurodegenerative disorders and track disease progression. Blood biomarkers are less invasive than CSF or PET but currently offer lower specificity. Translating biomarker findings into clinical practice therefore remains difficult1127.
Mechanism of Action: Anti-tau antibodies bind hyperphosphorylated, oligomeric, or aggregated tau, preventing its detachment from microtubules and subsequent destabilization74. They block aggregation of tau monomers/oligomers (Figure 2) into paired helical filaments (PHFs) and neurofibrillary tangles (NFTs)75. Fc-receptor engagement on microglia promotes phagocytosis of antibody–tau complexes, followed by lysosomal degradation, thereby reducing intra- and extracellular tau74. Restored tau homeostasis stabilizes microtubules, maintains axonal transport, and attenuates glia-mediated inflammation76. Anti-tau antibodies also preserve neuronal-membrane integrity by neutralizing toxic tau oligomers that disturb calcium balance and synaptic function75.
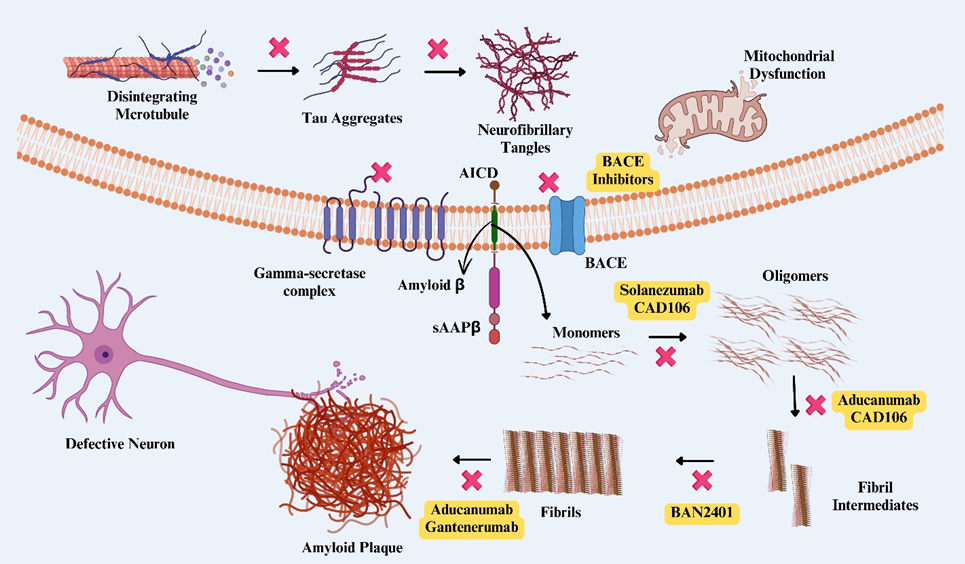
Formation of Aβ and tau tangles. Mechanism of action of drugs targeting amyloid plaques and tau tangles. 1. Drugs (in the yellow background) targeted at specific sites are under clinical trial, some of which are on the market, and some are discontinued. 2. ❌ indicates a possible site or target for the prevention or removal of plaque or tangles
Examples under investigation
• Semorinemab (Genentech/Roche) has shown mixed results; initial trials were inconclusive, but later studies suggest modest neuroprotective effects777879.
• Zagotenemab (LY3303560, Eli Lilly) demonstrated tau reduction and memory improvement in animals, yet early-stage human data remain inconclusive8081.
• Tilavonemab (ABBV-8E12, AbbVie), an IgG1-λ antibody, lowered tau pathology and showed encouraging safety/biomarker signals in Phase 1-2 studies8283. Trial M15-562 (Table 3) is a Phase 2, double-blind, placebo-controlled study evaluating low- and high-dose tilavonemab84858687.
|
Compound |
Trial (NCT No. / Phase) |
Trial Details (Phase / Duration / Participants) |
Limitations |
Controversy |
Statistical Results |
|
Semorinemab |
NCT03289143 (Phase 2 – Tauriel) |
Phase 2 / 73 weeks / 457 patients with mild AD |
No significant benefit on secondary cognitive/functional measures; limited sample size |
Targeting tau at symptomatic stage questioned; no dose-dependent efficacy |
Failed primary endpoint (CDR-SB, |
|
Zagotenemab |
NCT02754830 (Phase 1) |
Phase 1 / 2016–2018 / 110 mixed participants (healthy + MCI + AD) |
Small sample size; short duration; focus on safety, not efficacy |
Increase in tau not clinically significant; unclear target engagement |
Linear pharmacokinetics; dose-dependent increase in plasma tau; no PET amyloid or MRI changes. |
|
NCT03019536 (Phase 1) |
Phase 1 / 2017–2019 / 24 MCI-AD & mild AD patients |
Small size; short 16-week period; no efficacy data |
Debate continues due to Phase 2 failures |
Safe; linear PK; no significant biomarker changes; no clinical efficacy | |
|
NCT03518073 (Phase 2) |
Phase 2 / 2018–2021 / 285 early AD patients |
No clinical/biomarker benefit; insufficient dose response |
Plasma tau increased but not linked to outcome; tau-targeting still under debate |
iADRS ratio > 1 for both doses; no significant changes in PET tau, MRI, NfL | |
|
Tilavonemab |
M15-562 (Phase 2 – PSP) |
Phase 2 / 2016–2019 / 377 PSP patients aged 49–86 |
No improvement on clinical or quality-of-life measures |
Efficacy of tau-targeting in PSP questioned; halted for inefficacy |
No significant difference in PSPRS; 87.5% reported AEs, 25.5% had severe AEs; study terminated early |
|
AADvac1 |
NCT01850238 (Phase 1) |
Phase 1 / 2013–2015 / Mild to moderate AD patients |
Short duration; small sample; not designed for efficacy |
Active tau immunotherapy strategy under scrutiny |
Safe; immunogenic; antibody response observed; exploratory biomarker trends |
|
NCT02579252 (Phase 2 – ADAMANT) |
Phase 2 / 2016–2019 / Mild AD patients |
No significant cognitive/functional benefits; limited efficacy evidence |
Modest post-hoc subgroup findings require further confirmation |
| |
|
ACI-35 |
NCT04445831 (Phase 1b/2a) |
Phase 1b/2a / 2019–2023 / 57 MCI/mild AD patients (age 50–75) |
Final results pending; short-duration, individual-level data undisclosed |
Link between antibody response and clinical benefit remains unproven |
Interim: significant increase in anti-pTau IgG titers; no efficacy stats available; strong immunogenicity |
• AADvac1 is an active vaccine developing a self-immune response against pathological tau; Phase 1-2 studies revealed robust immunogenicity and tau reduction. Phase 3 is ongoing8485 (NCT01850238; NCT02579252).
• ACI-35, a liposome-based vaccine, elicited selective anti-tau immunity in Phase 1 and is now in Phase 2 for efficacy evaluation8687.
Combination Therapies and Novel Approaches
• Tau-aggregation inhibitor TRx0237 (LMTX) dissolves existing tangles and may halt or reverse dementia progression99.
• Neuroinflammation modulators such as sargramostim reduce immune dysregulation and may improve cognition; mefenamic acid is under study for similar effects100.
• NLRP3-inflammasome inhibitors (e.g., MCC950, tetramethylpyrazine, kakonein) decrease neuroinflammation in preclinical AD models101102103.
• β-Site amyloid precursor-protein cleaving enzyme-1 (BACE1) inhibitors (verubecestat, lanabecestat) lower Aβ synthesis by inhibiting β-secretase104. However, many agents (e.g., NB-360) were halted owing to adverse effects such as hair depigmentation, anxiety, weight loss, falls, suicidality, and sleep disorders. Umibecestat (CNP520) advanced further because of higher selectivity and favorable pharmacokinetics, yet overall risks highlight the difficulty of targeting amyloid pathways105106.
DRUG DELIVERY SYSTEMS TARGETING Aβ PLAQUES AND TAU TANGLES
Nanoparticle-based delivery systems
Polymeric Nanoparticles: Polymeric nanoparticles have been found to be useful for penetrating the blood–brain barrier (BBB) and releasing drugs at the target, i.e., Aβ plaques107108. Experiments have also shown that these nanoparticles can be functionalized with targeting ligands that increase their selectivity toward Aβ and therefore enhance the efficacy of drug delivery and decrease the amyloid load in the brain109110.
Lipid Nanoparticles: Lipid nanoparticles have potential in the encapsulation and distribution of therapeutic agents that deal with Aβ and tau aggregates111112. These systems offer long-term stability and controlled release, can cross the BBB, and may reduce neurotoxicity while improving cognitive function113.
Exosome-based delivery systems
Exosome Engineering: Small interfering RNAs and other small molecules can be incorporated into exosomes, naturally occurring vesicles 40–100 nm in size, to target neurons114. Exosomes can enter the brain and release their contents, specifically near Aβ plaques and tau tangles115.
Exosome-Loaded Drug Carriers: Exosomes combined with other delivery systems and nanoparticles can increase the specificity and efficiency of drug delivery. Preclinical studies of exosome-loaded nanoparticles have shown their potential to target Aβ and tau tangles116117.
Peptide-based delivery systems
Peptide-Conjugated Nanoparticles: Nanoparticles can be functionalized with high-affinity peptides that specifically bind Aβ plaques and tau tangles, resulting in enhanced targeting efficiency118. For therapeutic applications, these peptide-conjugated nanoparticles can effectively transport active agents, including antibodies and small molecules, to pathological regions to improve treatment outcomes and minimize adverse effects119120.
Cell-Penetrating Peptides (CPPs): CPPs are used to carry therapeutically valuable agents across cell membranes121. CPPs can be coupled with a drug or genetic material to improve their uptake by neurons, thereby directly targeting Aβ and tau damage and possibly even altering the disease course122.
Liposome-based delivery systems
Immunoliposomes: Immunoliposomes, which are liposomes linked with particular antibodies against Aβ or tau, have been created to enhance drug-delivery selectivity and effectiveness123. The BBB permits only regulated entry of substances, posing a major challenge to delivering Alzheimer’s-disease therapeutics into the brain. For large molecules like monoclonal antibodies (e.g., aducanumab), transport is restricted, and this requires an intravenous infusion or specialized methods of delivery. Small-molecule drugs generally have either low bioavailability or rapid clearance. Possible strategies include nanoparticle-based delivery, receptor-mediated transcytosis, and focused ultrasound for temporary opening of the BBB to maximize therapeutic effects and minimize systemic side effects. These systems can cross the BBB and thus provide ligands to affected regions; consequently, they have demonstrated their ability to reduce amyloid and tau accumulation in preclinical trials124125.
Multifunctional Liposomes: Liposomes with targeting ligands, imaging agents, and therapeutic agents are useful in the management of multiple aspects of this disease126. Liposomes can deliver and release drugs, monitor the response and effectiveness of treatment, and respond accurately to Aβ and tau tangles127128.
Limitations and Challenges in Drug-Delivery Systems
• Blood–Brain Barrier (BBB) Permeability: The BBB remains a major obstacle, limiting the ability of therapeutic agents (especially large molecules and biologics) to reach effective concentrations in the brain parenchyma. At the molecular level, this barrier limits transcytosis and receptor-mediated transport unless specific ligands or transport mechanisms are exploited129.
• Rapid Systemic Clearance: Nanoparticles, liposomes, and other delivery vehicles are often rapidly cleared by the reticuloendothelial system (RES), leading to reduced circulation time and poor central nervous system (CNS) bioavailability. This clearance depends on molecular surface features such as charge and hydrophilicity, which can reduce systemic circulation and CNS accumulation130.
• Immunogenicity and Biocompatibility: Synthetic carriers are recognized as foreign particles by the immune system, which may trigger inflammatory responses, immune clearance, or allergic reactions. Minor changes in surface chemistry at the molecular level (e.g., terminal groups, PEG density) can dramatically affect immune recognition131.
• Inconsistent Physicochemical Properties: Variability in nanoparticle synthesis can lead to differences in size, surface charge, and morphology, affecting drug-loading efficiency, release kinetics, and targeting accuracy. Batch-to-batch variability in nanoparticle synthesis (e.g., inconsistent nucleation or polymerization rates) can alter critical parameters like zeta potential, hydrodynamic diameter, and surface-ligand density, which influence molecular interactions with the BBB and target cells132.
• Potential toxicity of carriers: Some carrier materials or their degradation products (e.g., cationic polymers, metal-based NPs) may generate reactive oxygen species (ROS) or interfere with cellular organelles and enzymes, leading to molecular-level toxicity133.
• Enzymatic degradation of peptides: Therapeutic peptides are highly susceptible to proteolytic enzymes (e.g., peptidases, endopeptidases) in blood and tissues, leading to cleavage at specific amino-acid residues and a short systemic half-life. Structural modification (e.g., D-amino acids, PEGylation) is often needed to enhance stability134.
• Cell-Penetrating Peptides: CPPs like TAT allow drug entry via direct translocation or endocytosis, but they lack receptor specificity and are prone to enzymatic cleavage. Modifying CPPs with drugs can change their conformation, reducing efficiency and stability at the molecular level135.
• Exosome Production Challenges: Exosomes—natural nanocarriers—are heterogeneous in composition (lipids, proteins, RNA). Isolating them in a reproducible, scalable, and clinical-grade manner requires controlling the molecular makeup, including tetraspanins (e.g., CD63, CD81) and surface markers, which is technically difficult136.
In addition to the points mentioned above, other challenges in drug delivery include cargo heterogeneity in exosomes, limited stability and shelf-life of formulations, weak correlation between pathology clearance and cognitive benefit, poor translational value of preclinical models, and uncertain long-term safety of novel delivery systems. Further research is required to overcome these challenges and enhance the efficacy of CNS-targeted therapies137138.
CONCLUSION
In conclusion, Alzheimer’s-disease pathogenesis is associated with neuronal death, disruption of synaptic connections, and changes in cognitive ability. APP, upon degradation, produces Aβ, and these plaques cause oxidative stress and inflammation that affect neurons; they also interfere with normal neuronal function.
Research in AD has shown that there is a relationship between tau and Aβ pathologies, and these findings have revealed a significant contribution of these two proteins to the neurotoxicity that characterizes AD. Monoclonal antibodies such as aducanumab or lecanemab have shown potential in eradicating Aβ plaques efficiently. Because these antibodies can bind to aggregated forms of Aβ, enhance immunological processes, and activate microglia, they contribute to the removal of these aggregates. The main goals of tau-targeted therapy are to stop tau phosphorylation, diminish tau deposition, and stabilize microtubules via different ligands, such as epothilones and kinase inhibitors. Other immunotherapeutic techniques that promote the removal of tau proteins from the brain have also been developed and found useful. Similarly, drug-delivery methods involving the encapsulation of peptides and nanoparticles or loading into exosomes make it possible to increase the activity and specificity of the aforementioned therapeutics.
Overall, the above discussion reveals that the association between tau and Aβ pathologies is complex, indicating that the development of AD therapeutics requires multiple, complementary interventions targeting several pathological factors. Subsequent work will have to continue investigating the molecular relationships between tau and Aβ to determine how to design drugs that can effectively target these pathways. Nevertheless, current and future research aims to help those affected by this chronic disease achieve a better prognosis and higher quality of life, owing to advanced knowledge of the disease’s pathophysiology and the continuing refinement of therapeutic interventions.
Abbreviations
Aβ: Amyloid-β; ACh: Acetylcholine; AD: Alzheimer's Disease; AICD: APP Intracellular Domain; ApoE: Apolipoprotein E; APP: Amyloid Precursor Protein; ARIA-E: Amyloid-Related Imaging Abnormalities - Edema; ARIA-H: Amyloid-Related Imaging Abnormalities - Hemosiderin; ASOs: Antisense Oligonucleotides; BACE1: Beta-site APP Cleaving Enzyme 1; BBB: Blood-Brain Barrier; β-CTF: β-C-Terminal Fragment; CDK5: Cyclin-Dependent Kinase 5; CDR-SB: Clinical Dementia Rating-Sum of Boxes; CNS: Central Nervous System; CPPs: Cell-Penetrating Peptides; CSF: Cerebrospinal Fluid; Fab: Fragment, Antigen-Binding; Fc: Fragment Crystallizable; FcRs: Fragment Crystallizable Receptors; FDA: Food and Drug Administration; GSK-3: Glycogen Synthase Kinase-3; IgG1: Immunoglobulin G1; mAbs: Monoclonal Antibodies; MAP: Microtubule-Associated Protein; MAPK: Mitogen-Activated Protein Kinase; MAPT: Microtubule-Associated Protein Tau; MRI: Magnetic Resonance Imaging; NFTs: Neurofibrillary Tangles; NfL: Neurofilament Light Chain; NLRP3: NLR Family Pyrin Domain Containing 3; PET: Positron Emission Tomography; PHFs: Paired Helical Filaments; PP2A: Protein Phosphatase 2A; PTMs: Post-Translational Modifications; RES: Reticuloendothelial System; RNA: Ribonucleic Acid; ROS: Reactive Oxygen Species; sAPPβ: soluble APPβ; SFs: Straight Filaments; TLRs: Toll-like Receptors
Acknowledgments
The authors are grateful to Integral University, Lucknow, for providing the amenities, space, and resources for this work. The authors express their sincere gratitude to the Dean, Research and Development for the kind support (manuscript communication number: IU/R&D/2025-MCN0003365).
Author’s contributions
All the authors contributed to the study concepts and design data. The collection and main manuscript writing were performed by Pushpendra Soni, Samman, and Salman Khan; the data analysis was performed by Kuldeep Singh, Arun Kumar, Abdul Hafeez, and Suvaiv; and the data were reviewed by Syed Misbahul Hasan and Shom Prakash Kushwaha. All the authors approved the final manuscript.
Funding
None.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have not used generative AI (a type of artificial intelligence technology that can produce various types of content including text, imagery, audio and synthetic data. Examples include ChatGPT, NovelAI, Jasper AI, Rytr AI, DALL-E, etc.) and AI-assisted technologies in the writing process before submission.
Competing interests
The authors declare that they have no competing interests.